Statistical Mechanics
Covers origins of molecular partition function, then addresses translational, rotational, vibrational and electronic components.
Statistical Mechanics Notes
Boltzmann Factor

Physical quantity

Boltzmann Law:
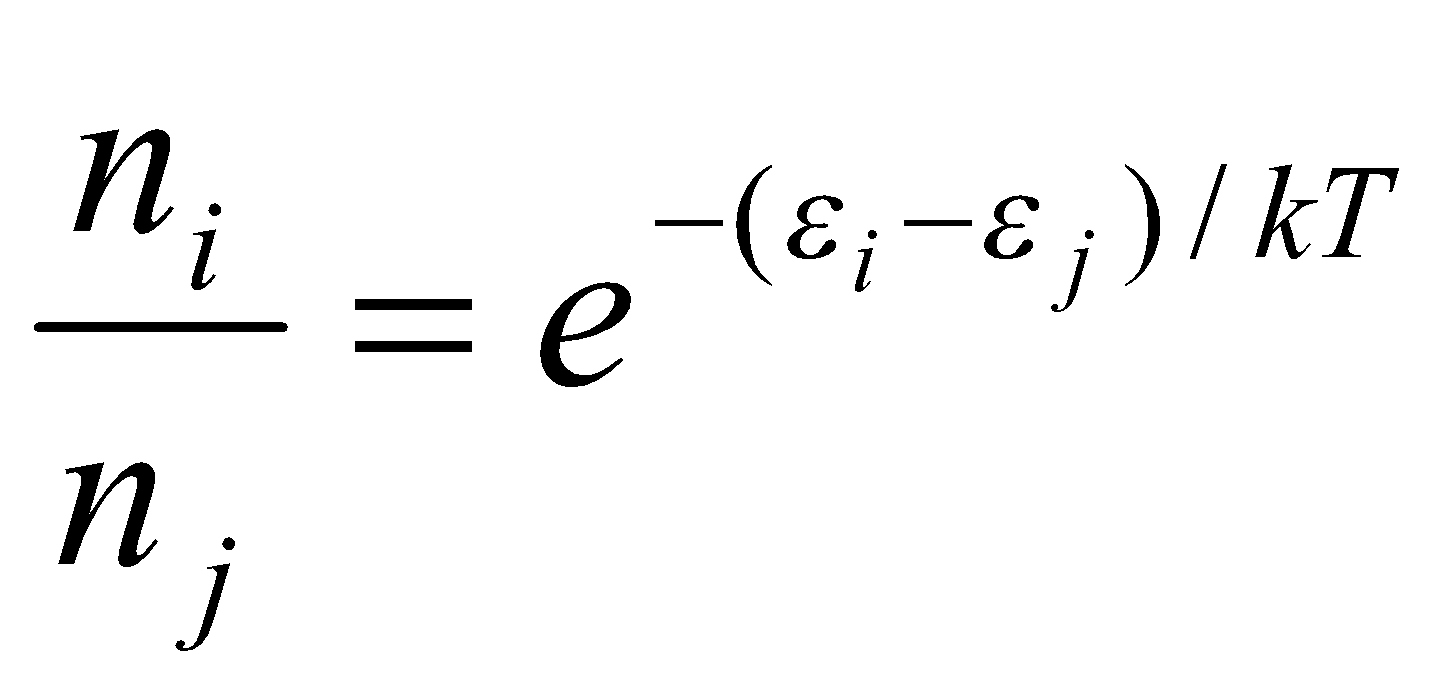
Distinct, Independent Particles –
Distinct → can tell which is which (labels) a,b,c…
Independent → minimal interaction (can exchange energy in collision).
Hence, E = εa + εb + εc + … =

Configurations –
Sharing energy amongst particles from a manifold of energy states, εo, ε1, ε2 … etc (ε0 = 0).
At any instant, there are:
- no particles with εo.
- n1 particles with ε1, etc.
This is the configuration. (Same Total Energy).
Statistical Weights –
Number of ways of reaching a given configuration, Ω. Represents the probability that the configuration can be reached.
, where x! = x(x-1)(x-2)(x-3)…3,2,1 and 0! = 1.
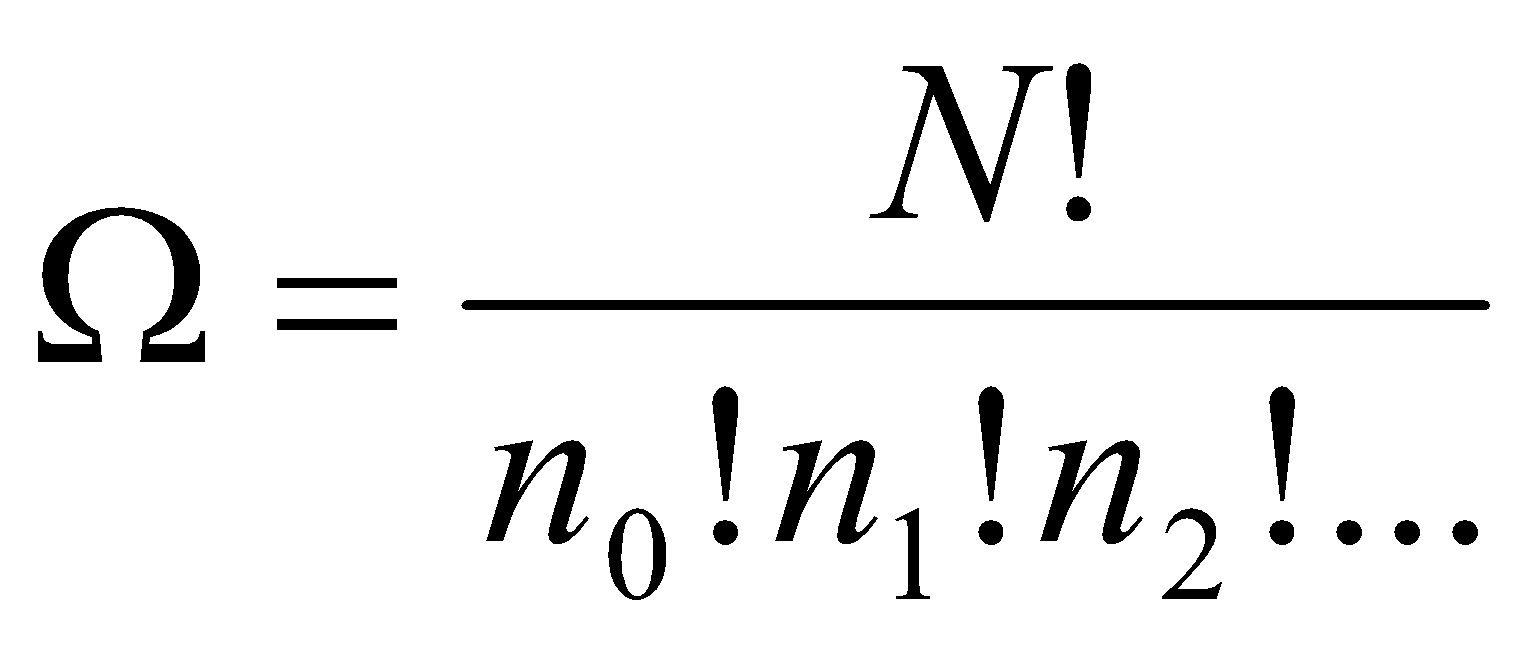
Equal Probability of Configurations –
No bias to any configuration. This is the Principle of equal a priori probabilities.
Conservation of Number and Energy –
&

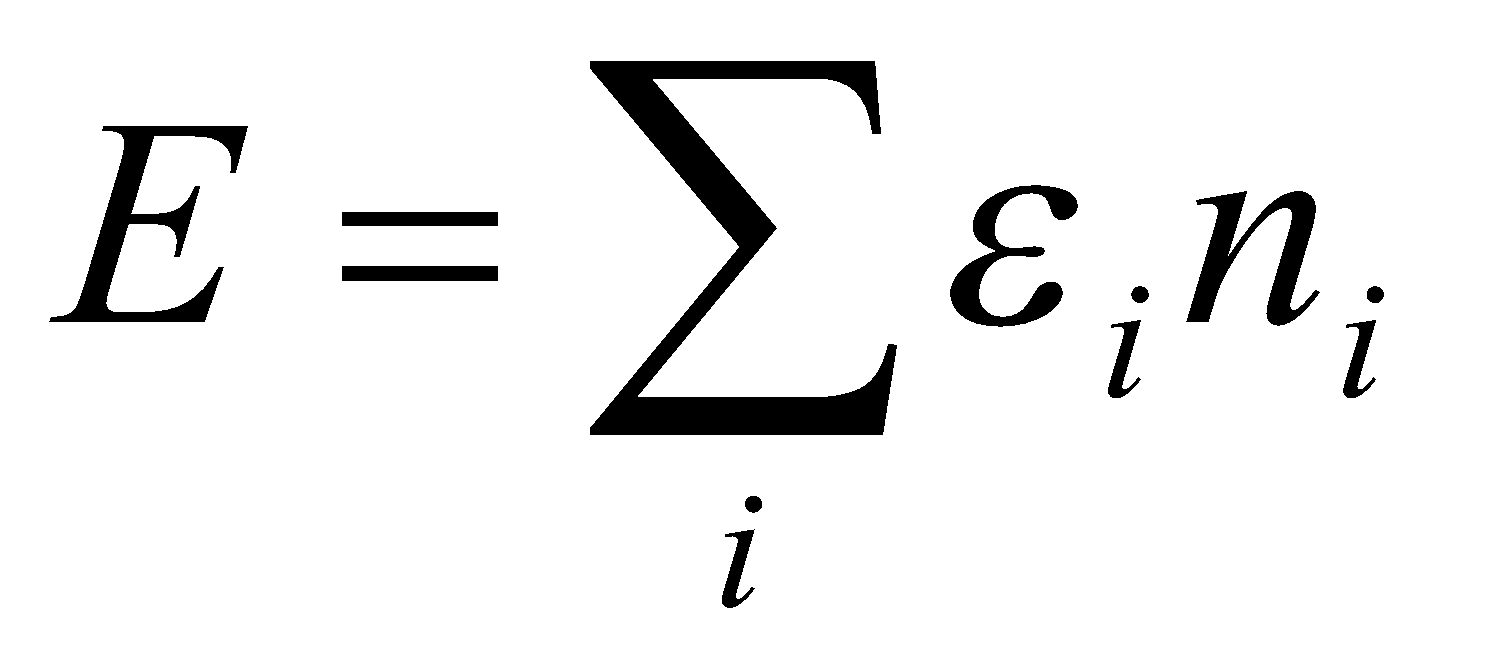
Predominant Configuration –
Configuration with the largest statistical weight.
For very large number of configurations, the average peak of the distribution completely dominates so that everything else is negligible.
Maximisation Subject to Constraints –
Find maximum in distribution (Ω) subject to constraints of Conservation.
Predominant configuration amongst N particles is found to have energy states populated as:
, where α and β are constants under fixed temperature.

Identify α:


This is the T dependent ratio.
β = 1/kT [ can be proven, see later ]
Molecular Partition Function
Derived from Boltzmann Law.

Eliminate no (generally not known):
N = no + n1 + n2 + … =

Hence,

Shows how particles distribute (partition) over accessible quantum states.
- Infinite series that converges more rapidly for increasing εi and increasing β.
- Can be evaluated as soon as βε >> 0, so that e-βε → 0.
- If ε1 >> kT, q → 1.
- For successive energy gap Δε, q >> 1 if Δε < kT.
Degeneracy –

Measure extent of particles escaping ground state.
T = 0K, q =1 (no = L).
Increasing T, q → ∞ (fewer particles in Ground State; infinite number of accessible states).
Applications –
Total Energy, E = n1ε1 + n2ε2 + … =
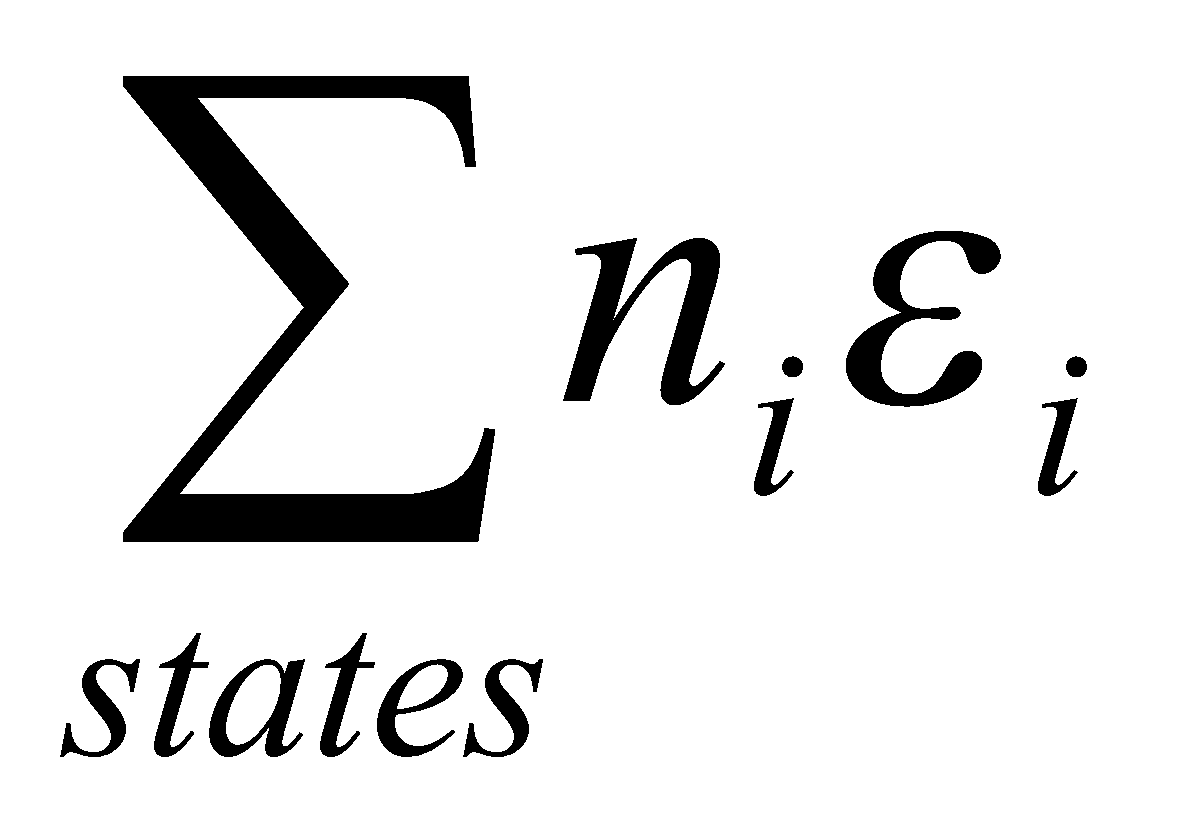
From q:

Note that:

Thus,
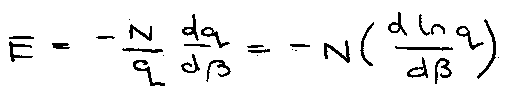
Internal Energy –
U = U(0) + E
U = U(0) - N

Also, q depends on V (which depends on T) so must specify constant volume:

This can be combined with the only temperature-dependent term in q for translational energy (see later):
ln q = -3/2 ln β
Such that:

This can be compared to N atoms in a perfect gas:
U = U(0) + 3/2NkT
And hence we see that β = 1/kT [ as used earlier ]
Entropy –
S = k ln Ω.
U =U(0) + E = U(0) +

At constant V (as for internal energy) the spacing of successive energy states does not change on heating. Thus, dεi = 0,
dU =

From here, dU = dqrev = T dS [ classical thermodynamics ]

Condition for max Ω (predominant configuration):

Number of particles is constant: = 0

Therefore,
dS = k d(ln Ω)
S = k ln Ω.
Molecule → Mole (Canonical Partition Function)

Reasonable when assuming non-interacting, and does not apply to other properties, e.g. S.
Allow possibility of interactions by invoking the idea that every system has a set of system energy states which molecules can populate.

Canonical Partition Function, QN :

Hence,

Compare to:

Can continue to develop a statistical toolkit of functions, e.g. Entropy:

Third Law → S0 = 0, on integrating:

Massieu Function
J = - A/T
This gives:

Links Statistical and Classical Thermodynamics.

Also, Pressure:

Heat Capacity,

Entropy:

Enthalpy:
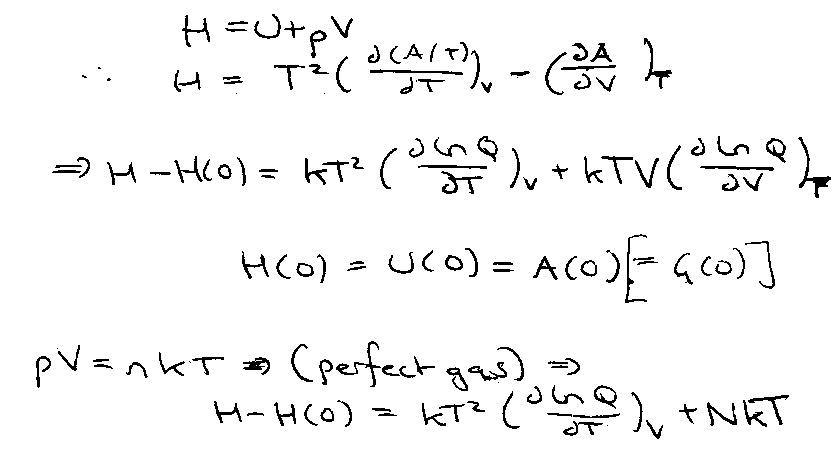
Gibbs Free Energy:
G = A + pV
G = A – V
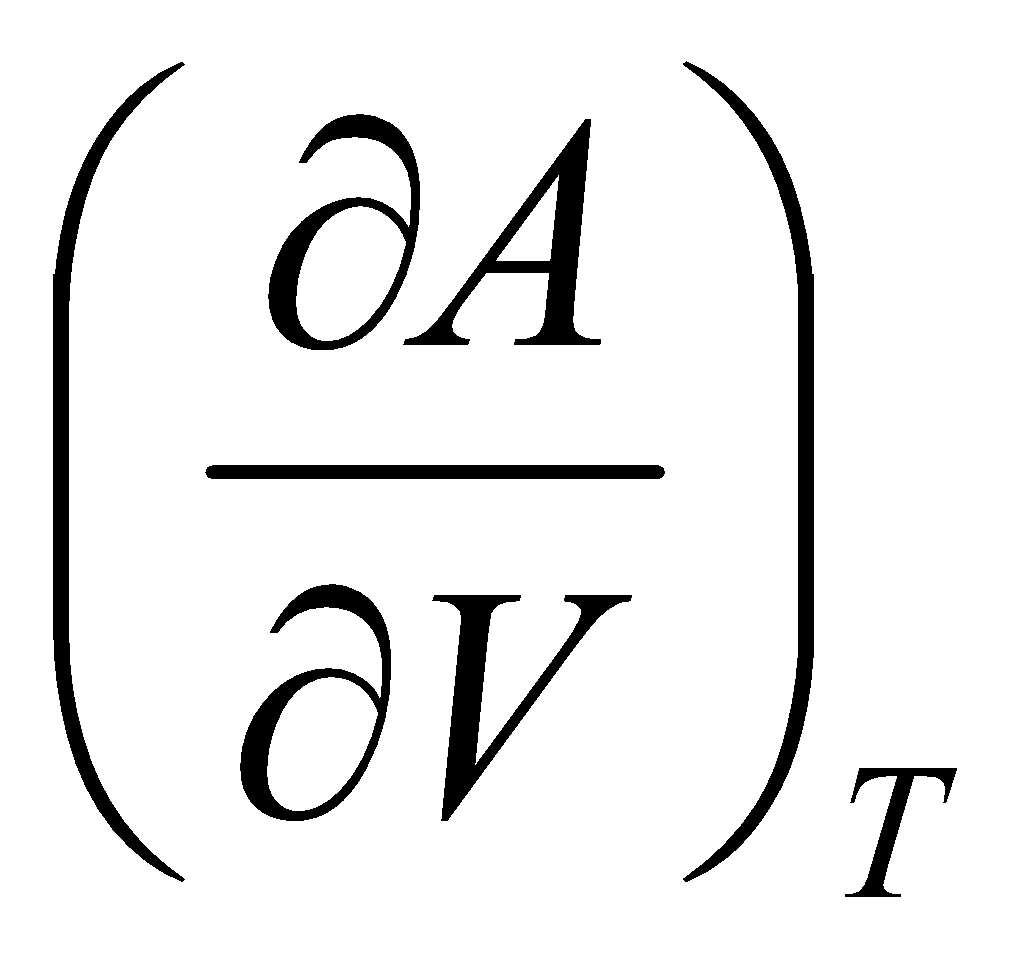

Perfect Gas →
G – G(0) = - kT ln Q + nkT.
Chemical Potential:

Independent Systems:
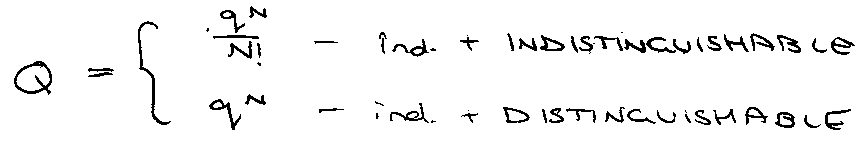
Use Stirling’s Approximation:
ln N! ≈ N ln N – N
e.g.
–kT ln Q = -NkT ln q + NkT ln N – NkT
Thus,
G – G(0) = -NkT ln (q/N) [ For INDISTINGUISHABLE ]
G – G(0) = -NkT ln q [ For DISTINGUISHABLE ]
Translational Partition Function, qtrs
Consider particle in a box:

Sum over all accessible states:

But practically all energy levels densely packed, so becomes an integral:

Canonically,

Collecting constants,
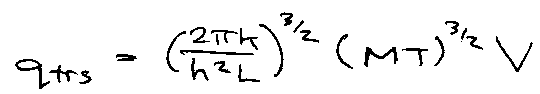
Thermodynamic Functions for an Ideal Monatomic Gas

Thus, derivatives simple:

These can then be used in the functions found earlier. Hence,

Entropy more complex, since ln Qtrs appears. It proceeds as:

Use Stirling’s Approximation: ln N! ≈ N ln N – N
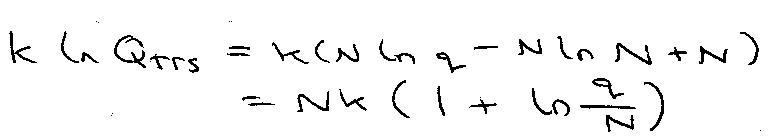
Thus,

Overall,

One mole of Ideal Gas →

This is the Sackur-Tetrode Equation. The constants add up to 172.29 J K-1 mol-1 [ 20.723R ]

Note that:
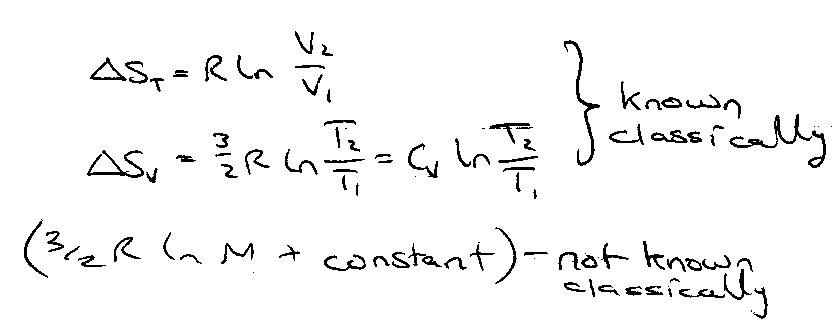
Ideal Diatomic Gas – Rotational Partition Function.
For a rigid rotor:


If I is not too small and T not too low, then appreciable number of rotational states are occupied, and there is a virtual continuum as for qtrs:

Problems tend to arise when T is nearly 0K and the molecule contains Hydrogen.
This expression otherwise works for all heteronuclear diatomics. Special considerations required for homonuclear diatomics. This is due to over-counting of rotational states by a factor of 2. This is because 180o rotation of X-X gives a result indistinguishable from 360o.
There qrot/2 required for all linear symmetric molecules.
Or,
qrot = T/σθr
Where:
σ = symmetry factor (= 2 for homonuclear diatomic, = 1 for heteronuclear diatomic).
σ = 2 for H2O and σ = 3 for NH3, for example.
Quantum Mechanically,
Interchange of identical nuclei may leave ψ unchanged, i.e. symmetric, or ψ → -ψ → antisymmetric.
Symmetric → boson (integral), while antisymmetric → fermion (half-integral).
ψtot =ψtrs.ψrot.ψvib.ψel.ψns
ψns = nuclear spin wavefunction. Symmetric or antisymmetric depending on whether 2 nuclear spin states are parallel / antiparallel.
ψrot = antisymmetric or symmetric. For even J it is symmetric, for odd J it is antisymmetric.
ψel = antisymmetric or symmetric. Homonuclear diatomic is usually 1Σg+ = symmetric. Only O2 is common exception, 3Σg- is antisymmetric.
ψtrs = only motion of centre of mass, so no effect on symmetry (symmetric).
ψvib = only depends on internuclear distance (symmetric).
Thus, in hydrogen I = ½ and the nucleus is a fermion so the ψtot is antisymmetric.
This requires odd J to give symmetric nuclear spin and even J to give antisymmetryic.
In Deuterium, I = 1 (boson) → ψtot is symmetric. Thus,
Odd J → paired nuclear spin.
Even J → parallel nuclear spin.
Thermodynamic Functions with qrot:
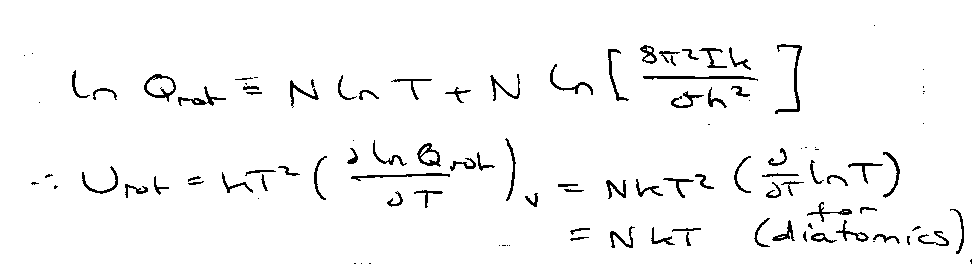
Applies to all linear molecules with only two degrees of freedom in rotation.
Molar → Urot = RT, Crot,m = R.
For entropy,

Extending to polyatomic, non-linear molecules – must consider 3 independent motions of inertia:

Ortho and Para Spin States
In general, for homonuclear diatomic with nuclear spin I, each nucleus have p (=2I+1) spin states, and a total of p2 nuclear spin wavefunctions to include in ψrot.
Of these p2,
½ p(p+1) = symmetric [ ORTHO ]
½ p(p-1) = antisymmetric [ PARA ]
This is true whether they are bosons or fermions.
Proton, I = ½ , ψtot = antisymmetric.
2 spin states, ↑ or ↓, therefore 4 ψns (p2)
Thus,
3 ortho (symmetric) = ODD J [ o-Hydrogen ]
1 para (antisymmetric) = EVEN J [ p-Hydrogen ]
Deuteron, I = 1, ψtot = symmetric.
3 spin states per nucleus, so 9 per molecule (p2)
6 x ortho (symmetric) = EVEN J [ o-Deuterium ]
3 x para (antisymmetric) = ODD J [ p-Deuterium ]
Therefore ortho-Hydrogen → Odd J, 3:1 ratio with odd predominating (due to statistical weight).
Similarly, ortho-Deuterium → Even J, 6:3 ratio with even predominating.
Ratios apply to e.g. rotational Raman Spectrum.
At high temperatures, hydrogen exists in an equilibrium mixture of spin states favouring o-Hydrogen by 3:1.
At low temperatures, there is an increased tendency for J = 0. Even → switch to p-Hydrogen. This conversion is slow.
Normal Hydrogen (n-H2) = 3:1 mixture.
Nuclei with zero spin –
Some nuclei are I=0 (e.g. 16O).
8 protons and 8 neutrons occupy their own energy manifolds (closed shell configurations – all spins paired). Also true for 12C.
p = 2I + 1 → O2 or CO2 have only 1 spin state.
I = 0 → integral, therefore boson, so ψtot = symmetric.
Hence,
CO2 → only even J.
O2 → only odd J.
Reason now is that ground electronic state is 3Σg- (i.e. antisymmetric), therefore rotation must also be antisymmetric (odd), unlike CO2.
Vibrations in an Ideal Diatomic, qvib
Orders of magnitude: qvib > qrot > qtrs are usually in the ratio 1:10:250-300 orders of magnitude. Thus, cannot use the continuum approximation for qvib.
qvib @ 300K ≈ 1.
Simple Harmonic –
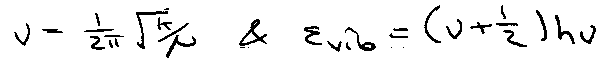
Always non-degenerate in diatomics. Not so for polyatomics – linear → (3N-5) normal vibrational modes, while non-linear → (3N-6).
εο = 0, ε1 = hv, ε2 = 2hv, etc, due to reference against ground state of ½hv. Thus,

This gives a geometric series:

This is true for diatomics only. For polyatomics, just consider each normal mode of vibration separately.
Vibrational Energy spacings are much larger than Rotational, therefore θvib ≈ 102-103, θrot ≈ 101-102.
Polyatomics –
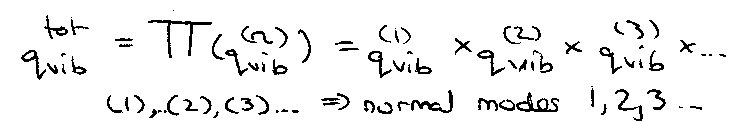
Independent and factorisable, therefore ignore anharmonicity (except at high T).
High Temperature Limit –
At high T, linear in qvib against T.
Expand :


Thermodynamic Functions –

Note: far less simple than Utrs and Urot.
At high T: Uvib,m = RT (for each normal mode)
At 300 K: Uvib,m =

Also note that if ε0 is set to ½hv instead, then must add this to the result.
Heat Capacity –
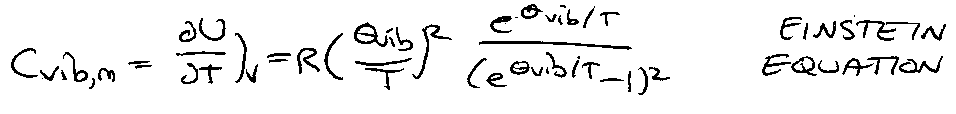

Entropy –

Electronic Partition Function

Ground States are commonly degenerate (not O2 though, g1 = 1).
For Atoms, use (2S+1)ΓJ and go = 2J+1.
For molecules, use (2S+1)Γ and go = 2S+1.
Excited States can be approached in a similar manner.
Usually, the energy gap from ground state to 1st excited state is large and the above applies. If the gap is not negligible compared to kT (i.e. θel/T << 1) then:

(Higher states than the 1st are rarely occupied).
I am happy for them to be reproduced, but please include credit to this website if you're putting them somewhere public please!