Review of the Periodic Table
Basically the lecture course delivered by a number of lecturers towards the end of third year. Good general revision material.
A Review of the Periodic Table
Atomic Structure and the Periodic Table
Solutions to Schrodinger Equation (orbitals) are usually expressed in terms of radial rather than Cartesian coordinates. The radial component can be plotted for various orbitals (dotted line). The Radial Distribution function, which gives the probability of finding an electron in a spherical shell between r and r+dr, is important for many electron atoms (shaded area):

The angular component of the solution to the Schrodinger Equation gives the shapes of the orbitals.
Many Electron Atoms
Each electron is regarded as moving in an average field – sum of nuclear field and that of other electrons. Effect of other electrons is to reduce the nuclear charge from the true value Z to Zeff.
The ability of electrons to reduce Z depends on the orbitals occupied, and the extent to which these penetrate towards the nucleus.
Shielding: ns > np > nd
More penetration leads to LOWER orbital energy.
Variation of Orbital Energies with increasing Z:

- The diagram above neglects electron correlation, i.e. change of energies of lower orbitals on ionisation.
- s-p separation actually increases across the period.
- s orbitals move into the core.
- By Zinc, 3d orbitals are low enough to become core orbitals.
At K and Ca, 4s is still lower than 3d, but small size of 3d causes greater sensitivity to increasing nuclear charge, and energy of 3d is lowered rapidly. Energy of the 4s decreases more slowly – hence the 3d transition series.


Energies of orbitals for non-hydrogen atoms given by:

The way in which orbitals fill (the Aufbau Principle) is governed by the energy sequences and Pauli Exclusion Principle.
Trends in Ionisation Energies
IE generally follows Zeff. Down the group, increasing IE outweighed by increase in ionic size. Anomalies across the first period due to exchange energy effects (compare N and O) or effects of filling a new shell (Be vs B), and then the variations for the 3d elements (see later).

The increase from K to Cu is due to the intervention of the 3d elements – 10 extra units of positive charge which are incompletely shielded by the 3d electrons. Likewise, for Cs to Au, the 4d and 4f are filled. Also note that there are relativistic effects which increase IE’s for the heavy elements.
Electron Affinities follow a similar pattern to IE’s, increasing left to right with anomalies at Group 15 (exchange energy). EA’s for the first row are lower than the 2nd row, due to smaller size (higher charge density).
Electronegativity
Pauling – larger energy of A-B bond cf. A-A and B-B due to electronegativity. Hence,
|χA − χB| = 0.102 x Δ0.5
Where Δ = E(A-B) – ½[E(A-A) + E(B-B)]
The Allred and Rochow definition is that electronegativity is a force experienced by valence electrons due to the nucleus. Zeff is then calculated using Slater’s Rules:
χAR = (3590 Zeff / r2) + 0.744.
The Mulliken scale is based on the fact that electronegativity relates to both IE and EA. Thus,
χM = (IE + EA) / 2
Note that the IE refers to the valence state of the combining atom and therefore includes promotion energies.
The trend seen is similar to those for IE, minus variations due to orbital occupation, exchange energy, etc.
- Increases across rows with increasing Zeff
- Decreases down group, but alternation effects
- Correlation with polarisability (Fajan’s Rules)
- Use of Ketelaar’s triangle to predict bonding type (see later).
Atomic Radii
- Across the period, atomic radius decreases as Zeff increases.
- Size increases down the group as increase in Zeff is outweighed by increase in n.
- Second and third row transition metals have similar radii due to Lanthanide Contraction, owing to low shielding by 4f electrons.
Ionic Radii
More complex picture than the above due to dependence on:
- Charge
- Coordination Number
- d electron configuration (for Transition Metals)
- Oxidation State (high = smaller radius).
The Shannon-Prewitt set of ionic radii are derived from X-ray structural data by assumption of O2- as 140 pm.
Thermochemical radii are used in the Kapustinskii Equation –
UL = 1.079x105 v z+z- / (r+ + r-)
Chemical Consequences of the Nucleus
Vibrational Spectroscopy
The mass of atoms are concentrated in the nuclei, and vibration frequencies are dependent on the mass. Isotopic substitution is thus useful for band assignment.
NMR
NMR active nuclei have spin angular momenta which can align parallel or opposed to applied magnetic fields. This gives rise to two nuclear energy states and higher can be populated by application of energy from radiation in the radiofrequency range. Transition is sensitive to nuclear surroundings – hence chemical shifts, etc.
Mossbauer
57Co 57*Fe 57Fe

e-capture gamma
Co source is moved towards and away from sample to shift frequency of gamma radiation from source and to cause absorption in same – the velocity is measured (isomer shift). Function of symmetry type of donor around Fe, oxidation state, etc.
Group 1 & 2 Metals
Group 1
Properties of Group 1 Elements:
|
ELEMENT |
IONIC RADIUS |
IONISATION ENERGY |
HEAT OF ATOMISATION |
MELTING POINT |
HEAT OF HYDRATION OF M+ |
ELECTRODE POTENTIAL |
|
Li |
0.6 |
526 |
161 |
181 |
-520 |
-3.0 |
|
Na |
0.95 |
502 |
109 |
98 |
-406 |
-2.7 |
|
K |
1.33 |
424 |
90 |
63 |
-320 |
-2.9 |
|
Rb |
1.48 |
409 |
86 |
39 |
-296 |
-3.0 |
|
Cs |
1.69 |
382 |
79 |
29 |
-264 |
-3.0 |
General trends follow Zeff – exception is non-systematic trends in E0.
Melting points low as only one electron available to delocalise in metallic state. All have BCC structures. E0 values can be calculated using a thermodynamic cycle:

- The dominant factor in E0 for Li is the hydration energy – higher for Li+ due to high charge density on small ion.
- Low value for an element such as Ag+ due to high heat of atomisation and high IE – latter due to high Zeff owing to filling of 3d and 4d shells.
General Comments:
- Chemistry is dominated by formation of monocations, high positive electrode potentials. Rapid reaction with water.
- Solid State structures archetypal of close packing model. Salts of the most polarising cations (Li+) and/or polarising anions have covalent character.
- High second IE ensures that M2+ compounds not stable. Use of Born-Haber cycle and Kapustinskii Equation shows that lattice energy is not sufficient to compensate for energy required to ionise second electron.
- Small size of Li induces some anomalies, however. It reacts with N2 to form Li3N, and carbon to form Li2C2. Nitrides of other Group 1 elements are unstable due to unfavourable lattice energy. Lithium Nitrate decomposes thermally direct to oxide, others form nitrite and O2 (UL also).
- Heating in air: Li gives Li2O, Na forms Na2O2, and K forms KO2. This is due to changes in lattice energy of solids with different anion size. High UL required to break O=O. Rb gives range of unusual oxides such as Rb6O and Rb9O2, which have metallic character, due to additional electrons from Rb not positively charged. Cs forms an even more extensive series, e.g. Cs3O11 and Cs7O.
- The balance of size and charge effects causes Li and Mg to have similarities in chemistry – “Diagonal Relationship”.
Crown Ethers and Cryptands
Crown Ether Ligands are of the type n-crown-m, where the ligand is an n-membered ring with m O atoms in it.
They form stable complexes with elements of Groups 1 and 2.
They have a lipophilic exterior, so are soluble in non-polar solvents.
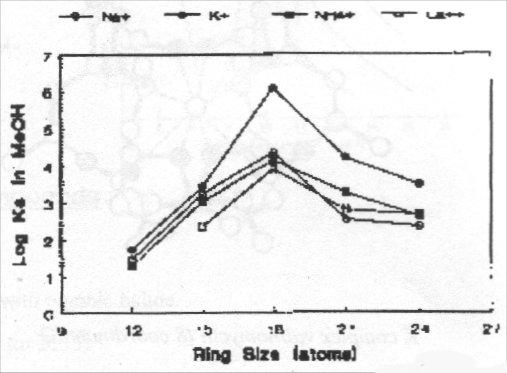
Plots of stability constant versus ring size for various Group 1 metal ions with 18-crown-6 shows clear maximum for K (right):
It is then possible to correlate stability with cavity size, but too simplistic - ΔH and ΔS terms important and M+ may not lie in plane of ligand.
Cryptand ligands encapsulate the metal ions completely, giving even higher stability constants. The K complex forms a capped trigonal prismatic structure:

There are many biologically occurring crown ethers and cryptands, which bind metal ions and transport them into cells.
Stabilisation of M-
If Na metal is treated with a cryptand or crown ether in a basic solvent, [Na(macrocycle)] + Na- is formed, and this can be crystallised. Na- ions are well separated from Na+, and formation of cryptate complex provides the thermodynamic driving force. Structure is essentially close packed [NaL]+ ions with Na- in octahedral holes.
Crystallisation from mixtures M and cryptand gives electrides ML++e-, with electrons occupying holes in a close packed lattice.
Solutions in Liquid Ammonia
- Liquid NH3 dissolves large amounts of Group 1 metals.
- Solutions have characteristic blue colour.
- Paramagnetic.
- Density LOWER than liquid NH3
- Contain solvated free electron, trapped in cavities in liquid NH3.
- Blue solution is a powerful reducing agent – reduces [Ni(CN)4]2- to [Ni(CN)4]4-.
Other Solvents
Solution of Group 1 metals in solvents such as TMEDA or THF produces a blue solution. However, UV/vis spectrum shows additional metal-dependent bands as well as that due to solvated electron. These are assigned to M-.
Organometallic Compounds
Organolithiums RLi are prepared by reaction of lithium metal with organic halide. Non-reactive solvents must be used, in an inert atmosphere.
They usually have polymeric structure in the solid state, but [(Me3Si)3C-Li-C(SiMe3)3]- is monomeric, and linear.
MeLi is tetrameric – a tetrahedron of Li with Me capping each of the four faces. Bonding is via 4c2e bonds. It is fluxional in solution, but the tetrahedral geometry is retained (exchange by Li-Li bond cleavage).
ButLi has the same structure. 7Li NMR (13C enriched) shows a septet – sum of spectra with coupling to 4 13C’s.
In the presence of donor solvents, they tend to break down into smaller units. PhLi with TMEDA is dimeric. Weaker donor solvent Et2O produces a tetrameric molecule. Phenyl carbon bridges 2 Li’s.
The high polarising power of Li+ causes partial negative charge on C – behaves as a source R-. Estimation of concentration via Gilman double titration, specific removal of RLi via reaction with CH2BrCH2Br.
Solubilities of Group 1 and 2 Compounds
Solubility of salts is governed by a number of factors:
- High dielectric constant facilitates dissolution by minimising coulombic interactions. Donor properties of solvent also important.
- Loss of lattice energy on solution compensates by heats of hydration of ions.
Heats of solution can be considered as thermodynamic cycle:

Salts with large mismatch of ionic size have relatively low lattice energy and high heats of hydration of small ion will cause such salts to be very soluble, e.g. CsCl. Insolubility of large cation-large anion due to low heats of solvation.
Above discussion neglects entropic effects:
- Strong ordering of solvent molecules around charged ions means entropy of solution frequently positive.
- For large anions such as NO3-, low UL and positive ΔSsol both increase solubility.
Free energies of solution for individual ions can be calculated for MX compounds, using Born Equation:
- Salts of small cations and anions are insoluble (LiF).
- Shows that for a given anion there is a minimum solubility – maximum in free energy for a certain cation radius.
Intercalation Compounds
- Direct reaction of graphite with K vapour gives C8K.
- K metal atoms inserted into alternate layers – increase in layer spacing, and graphite layers eclipsed rather than staggered.
- Heating under reduced pressure removes metal – C24M … C60M, with molten K, C4K, double layers.
- Resistance lower than graphite, and shows Temperature Independent Paramagnetism.
- React with MXn to insert M atoms between layers.
- Reaction between Group 1 metals and fullerenes gives MnC60 (e.g. K3C60).
- FCC structure with K in octahedral and tetrahedral sites.
- High Temperature Superconductors – Tc = 19.3K for K3C60.
- Electrons from M occupy π-orbitals on fullerene.
Group 2
- Trends in properties generally follow Zeff smoothly down the group, just like Group 1.
- Complications of changes in structure of metals are seen in the heats of atomisation.
- Also, the Chemistry of Beryllium is anomalous, and best treated separately.
Be Chemistry
Be2+ only known in the hydrated form, e.g. [Be(H2O)4][SO4], and high charge density causes ready formation of species such as [Be(OH)3]33+ by hydrolysis.
BeO dissolves in alkali to form [Be(OH)4]2- - beryllate ion, cf. Al, but not other Group 2.
Dominant coordination number is 4, although 2 and 3 known for large ligands (e.g. Be(OR)2) and in gas phase for BeCl2.
Most compounds are substantially covalent, e.g. BeH2 – synthesis by heating BeBut2 has polymeric chain structure with bridging hydrogen – 2e3c MO bonding scheme (like borane). Other Group 2 hydrides are ionic with salt-like character.
Unusual structures such as Be4OL6 (L = acetate, nitrate). Tetrahedron of Be with O at centre. Bidentate ligands span each of 6 edges. Each Be is tetrahedral.
Organometallics – like BeCl2, BeMe2 is polymeric in the solid state. Similar to Al2Me6 – shows diagonal relationship between Be and Al. Extremely toxic and reactive to air.
Cyclopentadienyl complexes also exist – CpBeCl and Cp2Be. In the former, the Cp ring is bound as η5, but in the latter both rings cannot be η5 as this would exceed 8 electrons around Be. Thus, a slipped structure forms with one ring essentially bound via one carbon – asymmetry is possible because of the small size of Be. Structures in the gas phase and solid are probably similar, and both rings appear equivalent by NMR (fluxional).
Mg, Sr, Ba, Ca Chemistry
General comments:
- IE values ensure that M3+ states are not accessible. Relatively high value for I2 is overcome by high lattice energies – M2+ occurs exclusively. Calculations via Born-Haber Cycles show that MgCl is stable wrt decomposition to its elements, but is unstable wrt disproportionation to MgCl2 and Mg.
- Greater ionic radius reduces charge density for cations and Mg aquo ion does not hydrolyse.
- Peroxides stable for Mg-Ba, not for Be. This is partly due to lattice energy, and partly due to high polarising power of Be2+ - destabilises peroxide anion.
Organometallics
Most important are Grignards RMgX.
- Prepare by reaction of Mg metal with RX in Et2O.
- Activation of Mg necessary. Catalysed by ultrasound.
- Rieke magnesium prepared by K reduction of MgCl2 – highly active.
- Estimation via Gilman titration.
Structure in solution more complex than suggested by simple formula:
- Several equilibria possible, dependent on solvent and nature of R. Halide bridges preferred.
- Polarisation by Mg causes carbanionic character for R, less reactive than RLi.
- Species present can be identified by 25Mg NMR.

- Magnesocene, Cp2Mg, has significant ionic contribution to bonding (evident from 13C NMR).
- Cp2Ca has an unusual polymeric structure.
- Cp*2M (M = Ca, Sr, Ba) is monomeric, with bent structures due to electrostatic effects.
Bioinorganic Chemistry of Groups 1 & 2
Cells have to maintain concentration differences for Na and K via highly specific ion channels.
Nerve impulses are created by a flow of Na or K cations across axon membranes.
Lithium carbonate used extensively as an anti-depressant.
Magnesium –
Chlorophyll – plays central part in photosynthesis. Contains Mg coordinated to porphyrin like molecule (corrin). Mg sits above plane of ring with water molecule(s) attached. Whole photosystem harvest light energy and transfers it to ferredoxin type molecules (reductants).
Role of Mg is to shift visible spectrum of free corrin and raise extinction coefficient of band by about 900nm. The water molecules bound to Mg form H-bonds which stack the Mg-corrin molecules to form a large array.
Phosphate Binding –
Polyphosphate groups bind Mg very strongly which catalyses the transfer of phosphate groups, and plays crucial role in enzymes such as ATPases, phosphotases and DNA polymerases.
Calcium –
- 99% of Ca is used for bone, teeth, etc. Bone is hydroxyapatite and turnover of 400mg per day of Ca in the body.
- Ca has crucial role as a trigger for biological processes such as muscle contraction where Ca2+ binds to a protein (troponin) causing a conformational change that in turn initiates muscle movement.
The Non-Metals
Ionisation Energies
- General trend is for an increase in IE as Zeff increases.
- Anomaly at Be due to start of filling p shell, that at O due to greater ease of removing a paired electron.
Looking at trend down e.g. Group 13 for sum of first 3 IE’s:
- Ga has higher IE than expected – Zeff increased as a consequence of filling of 3d which shield poorly.
- Similarly for Tl owing to filling of 4f shells.
This is the Alternation Effect.
Looking at Electron Affinities:
- Increase very markedly across 1st row. Anomaly at N reflects exchange energy effects.
- Suggests increasing importance for anions for non-metals and particularly to the right.

Electronegativity shows similar trend.
Cations or Anions or Covalent?
Whether cation or anion formed is a delicate balance of lattice energies, IE or EA. But compounds of non-metals usually covalent molecular species, so bond strength arguments are used.
Ketelaar’s Triangle can be used to predict type of compound formed between two elements:
Large electronegativity difference – IONIC.
Small electronegativity difference – COVALENT.
Homopolar Bonds
Tables below summarise ability of non-metals to bond to themselves (in kJ mol-1):
|
H-H |
436 |
|
|
|
|
|
|
|
|
|
B-B |
308 |
C-C |
347 |
N-N |
158 |
O-O |
144 |
F-F |
158 |
|
- |
- |
Si-Si |
226 |
P-P |
198 |
S-S |
266 |
Cl-Cl |
243 |
|
Ga-Ga |
113 |
Ge-Ge |
188 |
As-As |
178 |
Se-Se |
192 |
Br-Br |
193 |
|
In-In |
100 |
Sn-Sn |
146 |
Sb-Sb |
141 |
Te-Te |
126 |
I-I |
151 |
- Repulsions from core and non-bonding electrons reduce bond energy for 1st row, particularly N-N, O-O and F-F.
- Maximum in group for P-P and S-S.
- Bond Energy decreases down the group – contrast to Transition Metals. Contraction in s orbitals due to Zeff and relativistic effects reduces effective overlap.
For multiple bonds:
- Strong in first row due to good overlap.
- Decrease down group, as for E-E single bonds, but no max for second row.
Components of Multiple Bonding
- π-bonding component is found to be largest for the 1st row.
- Structures reflect balance between two types – for 1st row σ + π larger than 2σ, but reverse true for 2nd row.
Significance for Chemistry
Trends reflected in forms of element:
- C forms strong C-C and C=C and allotropes exist with C-C multiple bonding. Si as diamond type structure with Si-Si. Si=Si can only be stabilised kinetically by use of very bulky groups.
- e.g. (Mes)2Si=Si(Mes)2 – orange crystals, by photolysis of Mes2Si(SiMe3)2. Non-planar structure with twist angles 0-10o depending on R. 10% decrease in Si-Si compared to single bond. Corresponding Ge=Ge and Sn=Sn known, larger distortions from planar.
Group 15 –
- Strong multiple bonds and weak N-N – little catenation for N.
- P exists in many allotropic forms – all involving P-P catenation. White P contains P4 tetrahedra. Red P has chains with P-P bonds.
Group 16 –
- Multiple bonding only for O2, strong tendency for S to form S-S bonds as in S8.
- Many other S rings – neutral, and cations [S8]2+, latter has transannular S-S interaction. Anions [Sn]2- also by reduction. S42+ isoelectronic with S4N4 - similar planar structure.
Hydrides
Trends in E-H:
- E-H increase across period.
- E-H decrease down group.
Structures:
Boron is unique in forming a vast array of cluster hydrides. Simplest is B2H6 with 3c2e bonding for bridging H.
Reactions of diborane –

11B NMR of B3H82- shows 9 line spectrum due to coupling with 8 equivalent hydrogens.
Higher boranes are categorised as closo, nido, arachno, hypho, depending on number of vacant vertices in polyhedron.
Structures can be rationalised via Wade’s Rules:
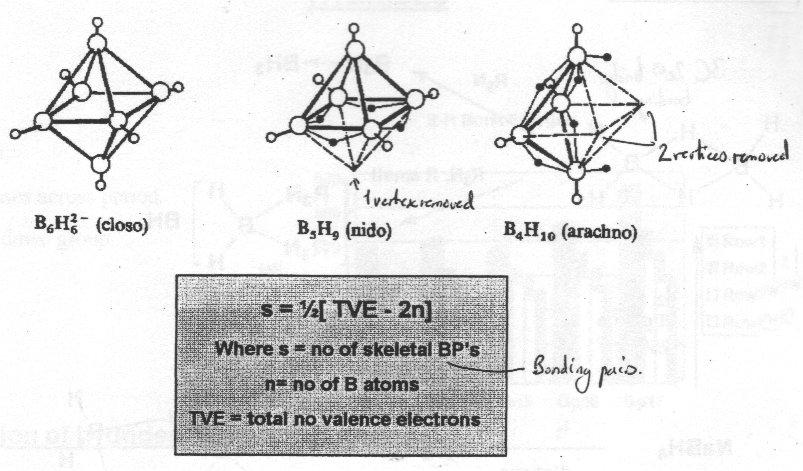
e.g. B5H9 – s = ½ [24-10] = 7 = n+2
s = n+1, closo.
s = n+2, nido.
s = n+3, arachno.
BH- can be replaced by CH to give extensive series of carboranes.
Zintyl Ions
These are homonuclear clusters of main group elements.
Synthesis: Element + Group 1 metal + crown ether in basic solvent, e.g. [Ge4]4-, [Pb5]2-, [Pb9]3-.
Also cations:
Bi + BiCl3 + AlCl3 → [Bi9]5+[AlCl4]5.
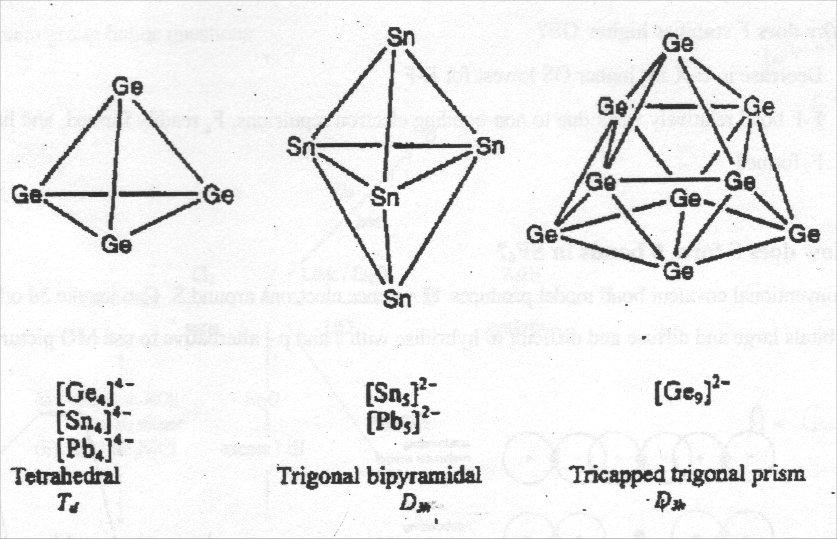
Structures can be interpreted using Wade’s Rules:
e.g. [Pb5]2- → s = ½ [20+2-10] = 6 = n+1 i.e. closo structure.
Note skeletal electron pair counting predicts capped square antiprism, actually tricapped trigonal prism, but energy differences between structures very small.
Hydrides of Group 14:
Few hydrides beyond Si due to instability. Hydrocarbons kinetically stabilised against reaction with O2.
Hydrides of Group 15:
N2H4 has gauche structure – rotated about N-N bond to minimise lone pair repulsions.
HNNH, diimide, unstable disproportionates to N2 and N2H4.
Halides
Trends in E-X:
- Bond Energy decreases across period: E-F > E-Cl > E-Br > E-I.
- For different oxidation numbers E-X decreases with increasing Oxidation State.
Why Does F stabilise higher Oxidation States?
Decrease in E-X for higher Oxidation States, lowest for E-F.
F-F bond relatively weak due to non-bonding electron repulsions, Fg readily formed, and little energy gain in F2 formed.
How does S form 6 bonds in SF6?
Conventional covalent bond model produces 12 valence electrons around S. Can invoke 3d orbitals, but d orbitals large and diffuse and difficult to hybridised with s and p – alternative to use MO picture:

F’s bound to S in three pairs, each comprising linear 3 centre MO formed by overlap of p orbitals.
These are filled by 4 electrons (2 from F, 2 from S) giving 3c4e bonding. d-orbitals may still be involved, but not essential.
Group 13 Halides
Acceptor ability of BX3 – BI3 > BBr3 > BCl3 > BF3 – reverse of order expected based on electronegativity. Due to π-bonding, greatest for BF3, which reduces availability of orbital on B to accept electrons.
Boron halides of type B2Cl4 by reduction of BCl3.
B4Cl4 are tetrameric, staggered structure in gas phase, but planar in solid state.
MX2 for Ga, is actually M+[MX4]-. But, addition of ligands gives X2LGa-GaLX2].
[GaCl3]2- also known, with a Ga-Ga bond shown by Raman Spectroscopy.
Note for Tl:
Tl2Cl3 is Tl(I)3[TlCl6]3- and [Tl2Cl9]2- has binuclear structure with triply bridging halides. TlI3 is not Tl(III) either, but Tl(I3).
Interhalogen Compounds
|
X-Y |
XY3 |
XY5 |
XY7 |
|
F-Cl |
F3Cl |
F5Br |
IF7 |
|
F-Br |
F3Br |
F5Cl |
|
|
|
F3I |
F5I |
|
|
|
Cl3I |
|
|
|
All 6 known Cl-F stable, I-F unstable wrt disproportionation |
IF3 unstable. Others volatile liquids, potent fluorinating agents. |
Volatile liquids. Powerful fluorinating agents, but less than ClF3 |
Low melting point solid. Planar bipyramid geometry. |
|
Shapes by VSEPR |
|||
Cations: by reactions with halide accepts, e.g. [ClF2]+[BF4]-
Anions: from halide donors, e.g. [Me4N][ClF4].
Polyiodide anions polymeric with I-I interactions.
Oxides, Oxoanions, and Sulphides
Chemistry tends to be dominated by formation of strong multiple bonds.
- Bond energy for these decrease down group and across period.
- E=O > E=S due to better overlap.
Metal Oxides
- Metal oxides are usually infinite lattices with close packed arrays of atoms.
- Metal has positive charge.
- Non-metal oxides are either molecular or polymeric with bridging O rather than E-E bonds.
- Tendency to form polymeric species increases going down the group, as E=O π-bonding component decreases vs. E-O.
- Decrease in heats of formation of oxides per oxygen across the table – corresponds to lower electronegativity difference, i.e. more covalent, more molecular.
- The oxides of Group 13 tend to be close packed structures (exception, B2O3 – polymeric).
- Groups 14-16 tend to be polymeric, although not true for those of C, N, O and S (all molecular).
- The halogen (and xenon) oxides are also molecular structures.
Some selected non-metal oxide structures:

- The chemistry in water is dominated by tendency for hydrolysis of ligated H2O.
- For non-metals capable of forming multiple bonds to O, oxo-anions are the most important species.
- Similar situation for metals that can form high oxidation states (e.g. Mn, Cr, Mo, Re, etc.). If metal cannot reach high oxidation states then polymeric species formed.
- Oxides of non-metals to top and right dissolve to give acid solutions.
- Redox characteristics of oxo-anions best followed by means of Frost Diagram.
- Polyoxoanions more oxidising with increasing numbers of O’s.
- More oxidising in acid.
- Acid strengths of oxo-acids can be predicted by Pauling’s Rules:
- For acid OpE(OH)n, pKa approximates to 8-5p
- Subsequent pKa values increase by 5 units for each proton lost.
Halogen Oxides

Sulphides
Allotropes – orthorhombic and β-monoclinic – S8 rings, different packing. Liquid S contains Sn chains. S-S bond flexible – 180-260pm.
Binary Metal Sulphides:
|
Type |
Elements |
Structure |
|
MS2 |
Li-Rb |
Anti-fluorite |
|
MS |
Mg-Ba |
NaCl |
|
MS |
Be, Zn-Hg |
ZnS (both types) |
|
MS |
Ti-Ni |
NiAs |
|
MS2 |
Ti-Hf, Pt, Sn |
CdI2 |
|
MS2 |
Mo, W |
Layered (MoS2) |
|
MS2 |
Fe, Ni |
Pyrites |
Polysulphides: S2-, Sn2-.
Important ligands for transition metals, particularly in biology (ferredoxins).
Fluorides:
S2F2, SF2, SSF2, SF4, SF5-, S2F10, SF6.
Cyclic N3S3X3 compounds – can be used to prepare M-NS complexes.
Oxides:
Controlled oxidation to S8O.
SO3(g) planar, tetrahedral solid state.
Nitrides
As with oxides, metallic nitrides have 3D lattice structures some of which have ionic character.
With non-metal covalent structures – molecular with first period, tendency for oligomers with later rows.
E-N bonds generally weaker than E-O as expected from electronegativity and high bond energy of N2 means that N compounds unstable wrt formation of gaseous N2.
Group 13:
BN has graphite type structure but with B-N interactions between layers – rings eclipsed.
Reaction of borane with NH3 gives B3N3H6 – delocalised structure with B-N multiple bonds. Arene type complex [(B3N3H6)Cr(CO)3] also known.
Group 14:
Chemistry limited beyond C, N(SiH3)3 planar due to steric effects and use of d orbitals on Si.
Group 15:
Phosphazenes (Cl2PN]n (n=3,4,etc) from PCl3 and NH4Cl.
Not delocalised like borazene - π-bonding islands localised on N’s which can be protonated. Compound with n=4 has boat conformation.
Group 16:
S4N4 prepared from S2Cl2 and NH3 or NH4Cl. Thermodynamically unstable wrt elements. Some S-N multiple bonding. Decomposes thermally in vacuo to give (SN)n polymeric chain structure – molecular wire.
At normal pressure S2N2 formed – cyclic planar 4-membered ring structure isoelectronic S42+.
NH isoelectronic with S, hence S7(NH) (cf. S8).
Group 17:
NF3 very weak base due to electron withdrawing effect of F. Very stable. By contrast NCl3 and NI3 very unstable. N2F4 also stable and exists as cis and trans isomers.
Organometallics Groups 13-15
Group 13
- BR3 type compounds – BX3 + 3LiR or NH3 + RCH=CH2.
- B(C6F5)3 (BARF) is a very strong non-coordinating Lewis Acid – used in olefin polymerisation reactions with Group 4 metallocene dialkyls.
- BPh4- - used to precipitate and stabilise large cationic metal species.
- Rings also known, e.g. Me6B3N3.
- Al2R6 type compounds – dimeric with 3c2e R bridges. Used extensively for Zeigler-Natta catalysis, particularly {MeAlO}n.
Group 14
- Extensive chemistry of polysilanes with Si-Si bonds.
- Si=Si stabilisation with bulky groups.
- For Sn, equilibrium between stannylene and species with Sn=Sn for R = CH(SiMe3)2:
2 SnCl2 + 4LiR → 2 :SnR2 ⇌ R2Sn=SnR2
- R4Sn stable.
- R4Pb used as antiknock agent – removes peroxides (as PbO) and terminates radical chain reactions.
Group 15
R3E type compounds: known for all of Group 15, ligating ability decreases down the group.
For E=P react with R’CH2X to give [RCH2PR3]+, then base gives ylids. Form olefins (Wittig).
R5E compounds for all of Group 15 – usually tbp geometry.
RP-PR kinetically stabilised by use of very large aryl groups.
Phosphaacetylenes accessible. Can act as metal ligands via P, and also cyclise to give P-containing macrocycles.
The Rare / Inert Gases (Group 18)
All elements have ns2np6 configuration.
Only interactions are Van der Waals – increase with increasing Z.
I1 less than that for O and F for heavier elements.
Fluorides – Structures:


Oxide Fluorides –

Does Xe+F- exist?
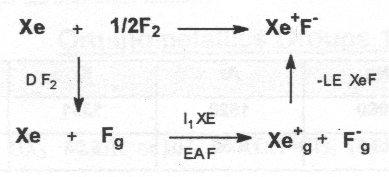
Consider the thermodynamic cycle – assume LE XeF approximately as for CsF, hence heat of formation +188kJ mol-1, i.e. not likely to form.
Cationic Fluoro Complexes
The fluorides react with F- abstractors such as [AsF6]- to give [XeF]+, [XeF5]+.
Latter sp structure in solution (19F NMR), but tetrameric in solid state with double F bridges.
Complex originally made by Bartlett and thought to be [Xe]+[PtF6]- actually mostly [XeF]+[PtF6]- with some [XeF]+[Pt2F11]-.
Anionic Fluoro Complexes
Complexes such as [XeF8]2- known.
129Xe NMR spectrum of [XeOF5]- has been shown.
Few Organometallics, only with C-F, e.g. [Xe(CF3)2].
Recent example of a radical cation with Xe-Xe bond:
[Xe-Xe]+[Sb2F11]-, with Xe-Xe = 308pm.
Formed from [XeF]+[Sb2F11]- with H2O under Xe, identified by EPR and Raman Spectroscopy.
Transition Metal Chemistry
Crystal Field Stabilisation Energy
If a TM ion is surrounded by 6 point negative charges at vertices of an octahedron, degeneracy of the five d orbitals is removed. Simple model ignores covalency effects.

- Tetrahedral splittings are smaller.
- Square planar picture explains stability of d8 square planar complexes, e.g. Rh(I).
- Possibility of high spin and low spin configurations – low spin is favoured by large splittings to overcome pairing energy.
- CFSE can be calculated for various electron configurations, e.g. d3 1.2Δ, low spin d6 2.4Δ. The diagram (right) shows CFSE for high spin octahedral and tetrahedral metal ions.
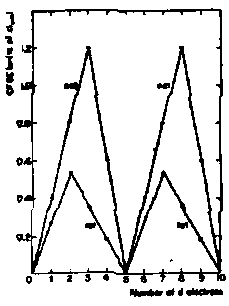
- Splitting magnitude dependent on metal charge (Fe3+ > Fe2+) and nature of ligands – spectrochemical series:
CO > CN- > PR3 > phen > bipy > en > NH3 > H2O > F- > Cl- > Br- > I-
- No simple explanation, but π-acceptors increase splitting overlap with t2g orbitals.
Alternative views of bonding via angular overlap model and MO theory.

Jahn-Teller Distortion
- Certain dn configurations are unstable wrt distortions that remove degeneracy.
- For octahedral, main examples are high spin d4 and d9.
- Figure for Cu(II) shows how distortion produces net stabilisation (right).
- Generally elongation along one axis (usually taken as z).
- Tetrahedral Cu(II) also distorted by flattening towards square planar.
First Row Transition Metals
In ground states, electron repulsion energies are minimised. For first row 4s is larger, occupied preferentially. Anomaly at d5 due to exchange energy effects (see below). On ionisation, energy of non-penetrating 3d orbitals drops rapidly while 4s is screened from increase in nuclear charge by 3d and fall slowly in energy – by Sc2+ 3d < 4s.
Net effect is that for TM ions, all valence electrons are d.
Ionic Radii
- Radii for octahedral low spin M3+ ions decrease as electrons are added to t2g orbitals, decreasing M-L distance.
- Start to increase again as electrons are added to eg. eg orbitals directed to ligands, and electrons repel negativity charged ligands.
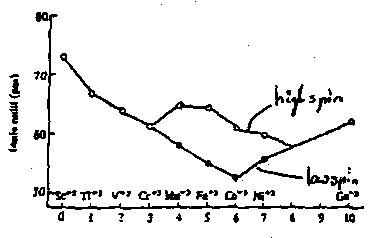
Heats of Hydration
- In absence of CFSE effects expect gradual increase with Zeff.
- Additional CFSE effect increases heats of hydration of M2+ - small compared to overall values.
- Influence of charge – M3+ > M2+


Rates of Substitution (Kinetic Effects of dn)
- Rates for exchange of coordinated water.
- Highly dependent on dn.
- Slow for d3, d6. Werner chemistry with Co(III) and Cr(III) as loss of CFSE in going to 5 coordinate intermediate required for Dissociative (Id) type mechanism or 7 coordinate intermediate for Associative (Ia) type. Mechanism changes from A at left hand end to D on the right (NMR evidence).
- Cr(II) and Cu(II) fast due to Jahn-Teller Effect (axial exchange faster).
- General decrease with increasing Zeff.
- As entropy increases, tendency for dissociative increases. Negative ΔV or ΔS suggests an associative mechanism – vacant metal orbitals.

Thermodynamic Stability of Complexes
- Trends in K1 and heats of formation follow pattern of CFSE values and ionic radii – Iving-Williams order.
- Dominant effect is change in Z*.
- More chelate effect makes en/EDTA favourable – entropically more favourable to bind to chelating ligand. EDTA also has coulombic forces.
- Low values of K5, K6 for Cu again due to Jahn-Teller.
- Co shows gradual decrease expected on statistical grounds.
- Cd(II) undergoes geometry change from octahedral to tetrahedral for 3rd I- ligand. Others 4 coordinate throughout.
- Fe(II) / phen system switches to low spin with 3rd phen ligand. Low spin d6 has higher CFSE than high spin.
Trends in Ionisation Energies for First Row Transition Metals
- For I1 + I2 maxima at d5 (Cr) and d10 (Cu) due to loss of exchange energy on ionisation of one or two electrons.
- For I3 anomaly now shifted to Mn.

Exchange Energy –
- Exchange Energy always negative and stabilising, opposite to repulsion effects.
- Plot of change in number of exchange stabilised electron pairs of electrons with parallel spins (Y axis, calculate from total number of possible combinations of pairs of electrons with parallel spins) versus number of d electrons shows maxima at d5 and d10.

Stabilities of MX2 and MX3
Construct a thermodynamic cycle for formation of MX3 from MX2.
Then, ΔHf = I3 – L(MX3) + L(MX2) +ΔHf(X-)


- L values show similar variation to heats of hydration – small fluctuations in comparison with I3, which is thus dominant.
- Plot shows free energy of overall reaction, very similar shape to I3. Small variation due to CFSE effects visible. MCl3 becomes less favoured to the right. MnCl3 less stable than FeCl3.
- Lower ΔHf(X-) for F- and effect on L makes ΔG much more negative – trivalent state favoured for MF3 – MnF3, and CoF3 stable.
Electrode Potentials
ΔH for redox process can be derived from a thermodynamic cycle as well:

Hence, ΔG = I3 + ΔHhyd(M3+) - ΔHhyd(M2+) – TΔS + ΔHH
- Plot shows trends in terms above, neglecting constant ΔHH term.
- Heats of hydration have been discussed. Note plot for M2+ inverted as positive term in the above equation – also smaller than M3+.
- Entropy term constant across the series.
- Hence, dominant term again is I3.
It is found that stability of M(II) increase to the right, while stability of M(III) correspondingly decreases (cf. halides).
Anomaly in I3 shows extra stability for Mn(II) (Mn(III) is oxidising).

Trends in redox potentials best displayed on a Frost Diagrams, which shows relatively stabilities:
- Decreasing stability left to right for M(II).
- All metals will give H2 in acid except Cu.
- Comparison with O2/H2O couple shows that Mn(III), Co(III) oxidise water (slope more positive).
- Ti(II) to Mn(II) oxidised by air to M(III).
- Mn(III) disproportionates to Mn(II) and Mn(IV).
- Cu(I) unstable wrt Cu and Cu(II).
- Many couples are pH dependent and less oxidising in alkali – high oxidation states more stable in basic solution.
Dependent of Redox Potential on Ligand
- As Δ value of ligands increase Co(III) becomes stabilised (by looking at E0 values for Co(II) / Co(III).
- For H2O, Co(III) reduced by water to Co(II).
- For ligands such as ox, phen, edta, NH3, increase in CFSE for low spin Co(III) stabilises Co(III).
- For Fe, π-acceptor ligands stabilise low oxidation state, so Fe(III) / Fe(II) for [Fe(phen)3]3+ is positive (i.e. Fe(II) favoured).
- σ-donor ligands such as EDTA stabilise Fe(III) – Fe(III)/Fe(II) now negative.
- Also, have to consider entropy effects caused mainly by hydration.
pH dependent of redox potentials is best displayed on a Pourbaix Diagram:
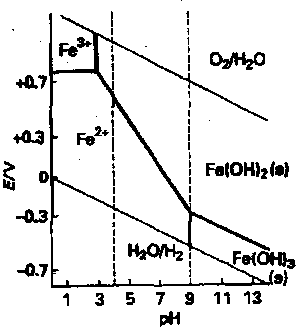
- Species to top oxidising agents, those at bottom reducing agents.
- Horizontal lines for redox independent of pH.
- Fe3+ only stable in solution at low pH and oxidising potentials, at higher pH Fe(OH)3 formed.
- Fe2+ has larger stability zone and exists under reducing conditions to pH 9 when Fe(OH)2 is formed.
- Sloping lines indicate pH dependent potentials.
Electron Transfer Between Complexes
Outer Sphere – Marcus model requires bond distances in complexes to change prior to electron transfer. Rates fastest for transfer of electron t2g to t2g or eg to eg. Rates calculated by Marcus Theory.
Inner Sphere – electron transfer via a bridging ligand. Three step process: bridge formation, electron transfer, dissociation. Rates affected by relative stabilities of intermediates (e.g. Co(III) vs. Co(II)).
Stable binuclear complexes with bridging ligands can transfer electrons between metals – Creutz-Taube ions. Binuclear complexes with metals in different Oxidation States can have three classes of delocalisation – none, complete, or intermediate.
Availability of Oxidation States
- First row show maximum oxidation state range in the centre of the series.
- Highest Oxidation States found with O and F.
- IE’s increase across row with increasing Zeff – higher Oxidation State not likely to the right.
- Too few electrons available to left to reach higher Oxidation States.
Sublimation Energies

- Metallic bonding highest at centre of series.
- But gain in exchange energy from delocalised metal to isolated atoms also highest for d5 – causes dip in centre.
- This term comes into play in considering heats of formation of MX2 from M and X2. Pattern for heats of formation MX2 will be superposition of those for heats of sublimation, lattice energies, and (I1 + I2).
2nd and 3rd Row Transition Metals
Atomic and Ionic Radii
- Decrease across series with Zeff.
- Lanthanide contraction causes similar radii for second and third rows – poor shielding by 4f causes 5d to feel higher charge.
Overlap and Electron Repulsion Effects
- 4d and 5d orbitals are more diffuse than 3d.
- Greater extension beyond core – better overlap with another metal or heteroatom. Hence M-M and M-X bond strengths increase down the group (contrast to p group where overlap decreases down group).
- More diffuse nature of 4d and 5d also reduces electron repulsion effects, so spin-pairing occurs more readily (exchange energy smaller). Also better overlap increases Δ, also favouring low spin states.
Ionisation Energies
- 1st few Ionisation Energies similar when comparing a 1st row metal with metal below it.
- Variation at higher IEs due to greater penetration of 4d and 5d which creates smaller increase in IE with larger positive charge.
- Has important effect that stabilities of higher oxidation states greater for second and third row.
Sublimation Energies
- Appreciably higher for 2nd and 3rd row – consistent with higher M-M bond strength.
- Less evidence for central dip due to exchange energy effects – reduced by larger size of 4d / 5d orbitals.

Metal-Metal Bonding
Increased ability of 2nd and 3rd row metals to form M-M bonds causes significant structural effects:
- For 1st row MCl2 compuonds these have octahedral M with bridging chloride and no M-M bonds.
- For MoCl2, valence electron deficiency is relieved by forming Mo6 octahedra in [Mo6Cl8Cl6]2- with 6 terminal Cl and 8 Cl located on the triangular faces bound to three Mo.
- Later metals can also form multiple M-M bonds (see below).
Redox Potentials
Data for Group 7 in acid solution:

Oxidising ability Mn > Tc > Re – similar for most groups. Reflects the IE differences. Despite formal similarity in chemistry of the 2nd and 3rd row, there are important differences.
M/Mn+ potentials are positive – reflects high heats of sublimation and high IE’s.
Lower oxidation states in aqueous solution more stable for 1st row. Simple hydrated M(II) and M(III) ions rare for 2nd and 3rd row.
Electronic Spectroscopy
Selection Rules
- Laporte rule – only transitions with change of parity (g→u, u→g) are permitted, i.e. d-d transitions are forbidden. Can be relaxed by unsymmetrical vibrations or d-p mixing in tetrahedral complexes.
- Change of spin state during transition forbidden.
For d1 cases transitions easily rationalised from simple CFT model.
For [Ti(H2O)6]3+ transition is from t2g to eg.
Shoulder due to J-T effect, as eg level split, giving two transitions.
With more than 1 d electron, electron-electron repulsions have to be considered.
For low values of Δ, effect of ligands perturbation of free ion terms (derived by Russell-Saunders coupling scheme) – weak field situation.
Situation where ligand field comparable with splitting of free ion terms – strong field case.
Now have to consider effects of ligands arranged in different symmetries.
How free ion terms split in octahedral and tetrahedral fields:

Dependence of energies of terms on crystal field displayed as an Orgel Diagram.
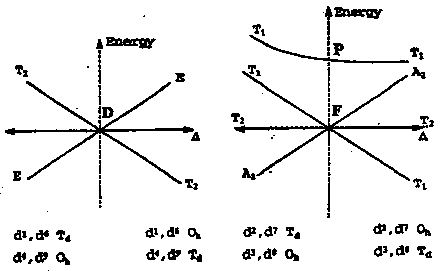
More quantitative picture provided by Tanabe-Sugano diagrams.

Plots E/B vs. Dq/B, where B is a Racah Parameter (energy separations of free ion terms can be expressed as multiples of Racah parameters B and C), and Dq is the crystal field splitting.
Ground State is now parallel to Dq/B axis, and all higher terms are shown.
If the energy at which absorptions occur is derived from spectra, then B and Dq can be determined from the Tanabe-Sugano diagram, and precise assignments made.
Charge Transfer Spectra
Diagram on right shows charge transfer transitions possible for metal complexes.
- Energies for transitions reflect ease of electron transfer, therefore correlate with IE of donor and EA of acceptor also with redox properties.
- Transitions are fully allowed, therefore very intense.
- Energy of transition for LMCT for [MO4]2- in the sequence Cr < Mo [Mo(VI) harder to reduce than Cr(VI)]. Also V(V) > Cr(VI).
- Similar sequences for [NiX4]2-. Energy of transition Cl > Br > I – follows reducing power of halide anion.
Magnetic Properties
Four classes of magnetic behaviour – diamagnetic, paramagnetic, ferromagnetic and antiferromagnetic – focus here on paramagnetism.
Molar susceptibility χM related to magnetic moment by expression

This is from Curie’s Law.
Rearranging and providing numerical value for constants, get an expression for the magnetic moment μeff:

For ions of first row metals:

Neglecting the orbital contribution (L=0) and substituting n = 2S, the spin-only formula is reached, where n = number of unpaired electrons:
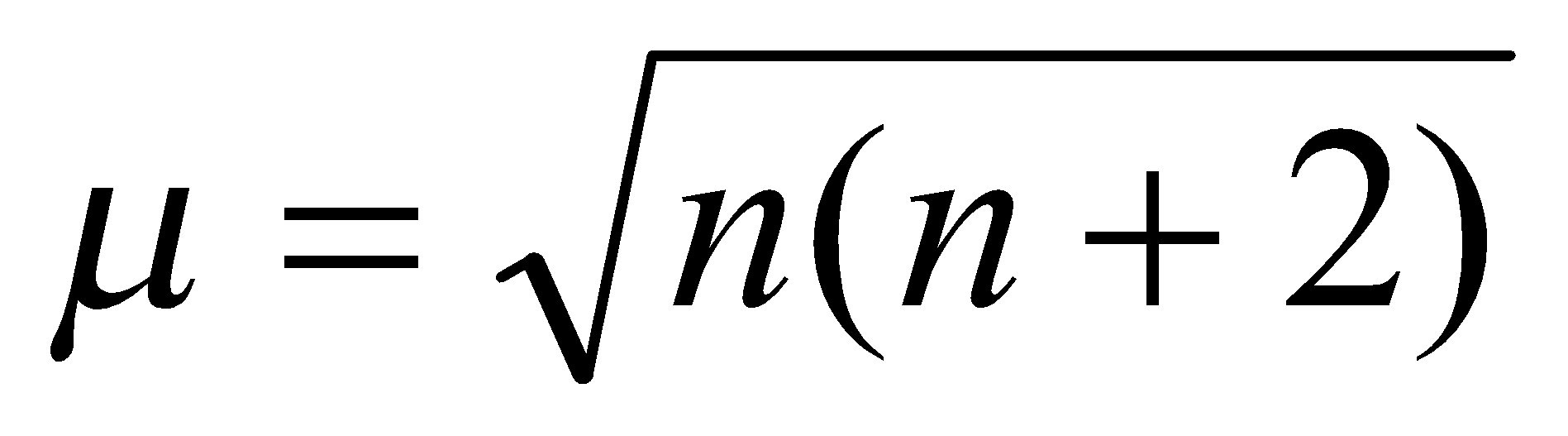
This assumes that the orbital contributions are quenched by the ligand fields. This is not true for complexes with T ground states, and for A and E ground states spin-orbit coupling introduces small but significant orbital contribution.
Complexes with A and E Ground States
Orbital contribution by second order spin-orbit coupling to excited states with same multiplicity gives the modified expression for the magnetic moment:

α = 2 for E terms, 4 for A terms. Positive for 1-4 electrons, negative for 6-9. λ is the spin-orbit coupling constant, and Δ the crystal field splitting.
Also a term from second order Zeeman Effect (Temperature Independent Paramagnetic term), but this is generally small and can be neglected. Use of this expression gives reasonable agreement between calculated and experimental values.
Complexes with T Ground States
These complexes have incompletely filled t2g orbitals, and there is an orbital contribution. Spin-orbit coupling is of the order of kT, and thermal distribution over these states causes temperature dependence – magnetic moment increases with temperature, diagnostic of T ground states. Quantification of effect on μ is difficult, but generally higher than spin-only value.
Magnetic moment with depend on relative magnitudes of spin-orbit coupling constant λ and kT – displayed as plots of μ vs. kT/λ - Kotani Diagrams.
Change of Spin State
If crystal field close to high spin / low spin switching point, then thermal equilibrium between two spin states. e.g. for Fe(II) sometimes get a change of 4 in number of unpaired electrons when moving between two temperatures near the crossover point.
Structure of the complex remains constant however – contrast to temperature dependence of μ for Ni complexes due to tetrahedral vs. square planar geometry change.
Metal-Metal Bonds and Clusters
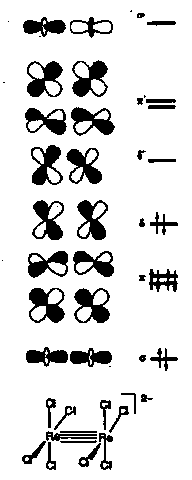
The widespread ability of transition metals to form M-M bonds is an integral feature of their chemistry.
MO scheme shows bonding for 2 ReCl4 units.
Geometry is eclipsed D4h.
If geometry is staggered then δ orbitals degenerate which can affect magnetic properties of the complex.
Possibility of quadruple bond – one σ bond, two π bonds and one δ bond, last unique to transition metals with d orbitals.
8 electrons required for quadruple bond (each metal d4, Re(III)). For later metals with π-acceptor ligands 34 valence electrons correspond to a single M-M bond, 32 to double, 30 to triple.
M-M multiple bonds can be supported by bridging ligands.
Other common examples of M-M quadruple bonds – [Mo2Cl8]4- and [Cr2(acetate)4]. Note different structure of last compound from CrCl2, which has octahedral Cr’s with bridging halides.
Carbonyl Clusters
- For small M-CO clusters, valence electron counting can be used to determine no M-M bonds.
- For [M3(CO)12] (M = Fe, Ru, Os), VE = 3x8 + 24 = 48, 3 M-M bonds (6e) required to make up to 54 electrons (for 18 electron rule).
- This does no predict the number of bridging CO’s – decrease down the group with increasing size and separation of metals.


- For larger clusters with increasing delocalisation, e.g. [Rh6(CO)16], this method fails.
- It is also difficult to deal with interstitial atoms, e.g. C, N, H.
- Use Wade’s Rules to determine polyhedral structure:
- s = ½ [TVE – 12n] where n = number of metal atoms, s = n+1 for closo, n+2 for nido, s = n for one cap, s = n-1 for two caps, etc.
- e.g. [Rh6(CO)16]: s = 0.5 [54+32-72] = 7 = n+1, i.e. closo octahedron.
- For even larger clusters (n>10) structure based on close packing of metals – transition to metallic state.
Isolobal relationships can also be useful in determining cluster structures containing both metals and non-metals. Two fragments are isolobal if number, symmetry and shape of orbitals and number of electrons in them similar.
Heteroelement-metal Multiple Bonds
Metal oxo-complexes:
- Found for high oxidation state of metals, e.g. M(IV) to M(VIII).
- Electron deficiency of metal relieved by donation of non-bonding electron pairs into appropriate M orbitals – also increase M-O bond strength.
- Transfer of oxo-groups important function of Mo in sulphate reductase.
- For d0 dioxo-complexes O groups usually cis to minimise competition for M orbitals, but UO22+ trans due to f-orbital involvement.
- Variety of bridging modes, geometry of single O dependent on π-donation.
- Alkoxides coordinate similarly – for electron deficient early metals strong tendency to bridge using available lone pairs. If R bulk then no bridging and MOR system can even be linear as both lone pairs donated to metal.
Metal-carbon multiple bonds:
M-C multiple bonds for CO achieved by synergic σ-π donation.
- M-CO has donation from filled metal orbitals into anti-bonding orbitals on CO – reduction of CO bond strength (IR and X-ray). Also polarisation of CO enhanced C(δ+)-O(δ-) – hence nucleophilic attack at C, electrophilic at O.
- Carbyne ligand has 1 σ and 2 π bonds.

Carbene complexes of two types:
- Fischer carbene with heteroatom at C – carbon δ+, attack by nucleophiles at C.
- Schrock carbene (alkylidene), no heteroatom – carbon δ-, attack by electrophiles at C.
- Alkylidenes important intermediates in olefin metathesis – formation of metallacyclobutane by reaction with olefin.
Metal-Nitrogen Multiple Bonds:
Typical species shown on right.

- Nitride: one σ and 2 π bonds – 3- charge, normally with high metal oxidation state. Strong bond important to provide thermodynamic driving force for reduction of N2 on metals.
- Imide: isoelectronic with oxo. Exists in linear or bent forms, depending on whether lone pair on N is donated to metal.
- NO can also bind in two analogous ways to metals – EAN rule can be used to predict bonding type in most but not all cases.
Ligand Perspective
Steric Effects –
- Introduction of bulky ligands – stabilisation of low coordination numbers / unusual geometries.
- For ligands such as RO-, RS-, steric bulk also minimises tendency to bridge.
- Electron deficiency of metals reduced by π-donation of lone pairs from N or S. Agostic interactions with C-H bonds also found for low coordination number complexes.
- Steric hindrance also offers kinetic stabilisation by preventing access to reactive metal centres.
- e.g. stabilisation of Mo-Mo triple bond using bulk alkoxides. Smaller ROH give OR bridged polymers.
- For tertiary phosphine ligands, steric effects rationalised by Tolman cone angle. Cone which touches metal and van der Waals surfaces of H atoms on ligand. Values from 118o for PMe3 to 194o for P(2,4,6-Me3C6H2)3. Donor ability also changing – indicated by pKa values.
- Polydentate ligands can also impose specific geometries on metals, e.g. Triamido(3-) ligand uses bulk and specific shape to impose unusual tbp geometry on Mo, and stabilises an N2 bridging ligand.
There are many examples where a metal is used to control formation of macrocyclic ligands:

Bonding and Activation of Dinitrogen
Group 4 –
First examples of N2 complexes all involved Cp ligands:
Cp2TiCl2 (+ Zn) → Cp2TiCl (+ PhMgBr) → Cp2TiPh (+ N2) → Cp2PhTi-NN-TiCp2Ph
Ti(III) complex diamagnetic due to antiferromagnetic exchange across N2 bridge.
For Ti(II) ‘titanocene’ chemistry complicated by CH activation formation of e.g. fulvalene derivatives.
Group 5 –
Despite involvement of V in certain types of nitrogenase, V N2 complexes are rare. They can be of the V(0) phosphine type – 17e count for V, prepared by reduction of V(III) in the presence of diphosphine and N2.
For Nb,Ta – bridged N2 complexes by metathesis of alkylidene complex with azine.
Have short Ta-N distance, and long N-N.
Group 6 –
Intense interact due to involvement of Mo at the active site of nitrogenase.
First complexes all M(0), 18 electron systems with PR3 ligands dominant.
Cis or trans depending on whether monodentate or bidentate. Larger PR3 only bind 3 P ligands and [M(N2)3P3] – max number of terminal N2 ligands bound to M under normal conditions. N2 is both a weaker π-acceptor and σ-donor than CO.
Much more recently, have been Mo and W complexes with triamide type ligands.
Group 7 –
Most of the chemistry of N2 has involved electrophilic attack at β-N. Alternative is nucleophilic attack at α-N – only possible for relatively electron deficient systems. Clear analogy with Fischer carbene synthesis from [M(CO)6] and RLi.
Also extensive chemistry of Re complexes of the type [ReCl(N2)(PR3)4] and analogues of Mn complex opposite.
A few general comments on the Coordination Chemistry of Dinitrogen –
- Bonding to metals analogous to CO, but N2 weaker π-acceptor and σ-donor than CO, no M(N2)6 except in matrices at very low T. M-N bond strength less than M-C hence use as labile leaving group.
- Pattern of occurrence of N2 complexes similar to that of OC. Few to right where energy of d-orbitals has increased and synergic bonding not possible. To the left Cp ligands required to increase effective electron density at the metal.
- Major difference to CO chemistry is the formation of M=N-N=M systems or “side on” bridging mode. Only possible for early metals with vacant orbitals and N can act as π-donor.
- If metal has no acceptor orbitals then M-N=N-M type bonding. Types of bridging mode can be distinguished by combination of Raman/IR.
- Synthesis of N2 complexes directly from N2 often difficult due to low solubility of N2 in organic solvents at RT (use pressure, low T) and that vacant sites are often lost due to competing reactions (formation of M-M bond, inclusion of solvent, formation of ligand bridged oligomers). Indirect methods such as Schrock Nb synthesis which proceeds presumably via 4-centre transition state, mean that N-N ligand always bound to metal. Stability of N2complexes often depends on steric protection of the M-N2 system from competing reactions as outlined above.
- The active site of the enzyme nitrogenase has been crystallographically characterised and shown to be a complex cluster of 7 Fe and 1 Mo with S bridges. Still a matter of dispute if the N2 is bound within the Fe-S cage or by Mo.
Reactivity
- For terminal N2 complexes polarisation is vital for electrophilic attack to occur (at β N). To the right of the periodic table the metal has greater electron density than the bound N2, and protonation at the metal occurs. The consequent increase in oxidation state leads to the loss of the N2.
- The β N of a terminally bound N2 ligand has a comparable basicity to MeCN towards a Lewis Acid such as AlEt3.
- Basicity is greater for W than Mo – reflects amount of 5d orbital outside core due to shielding and indirect relativistic effects. W is then able to transfer more electron density to the N2. Also shown in fact that N2 is bound more strong to W = for [M(N2)2(diphosphine)2] N2 loss occurs at RT for Mo, but photolysis required for W.
- Although O is more electronegative than N, M-N2 generally more easily protonated than O of M-CO (occurs in carbonyl clusters for a few CO complexes only). CO can in principle be protonated once to give M≡C-OH whereas N2 protonates readily twice to give stable M≡N-NH2.
- For bridged N2 complexes protonation occurs to give M-NH-NH-M intermediates.
Metal Hydrides

Interaction of H2 with Metals –
Cleavage of H-H bond.
Favoured by relatively e-rich metal centres.
For less e-rich metals η2-(H-H) complexes possible.
Identification of Hydrides
High field shift of NMR (higher paramagnetic contribution to the shielding).
For η2-hydrides T1 (longitudinal relaxation time) is reduced . If T1 < 40ms the dihydrogen complex, if >100ms definitely dihydride.
IR spectra show M-H for hydrides 1700-2000cm-1. Dihydrogen complexes may show H-H at 2500-3100cm-1 (cf. 4000cm-1 for H2 itself).
Neutron Diffraction used to locate hydrogens, but large crystals needed. X-ray structures show possible locations of hydrides.
[PtClH(PR3)2] –
Prepared by reaction of dichloride with KOH/EtOH as a white square planar air stable solid.
Pt-H appears in the IR at about 2000cm-1. NMR most useful for identification:
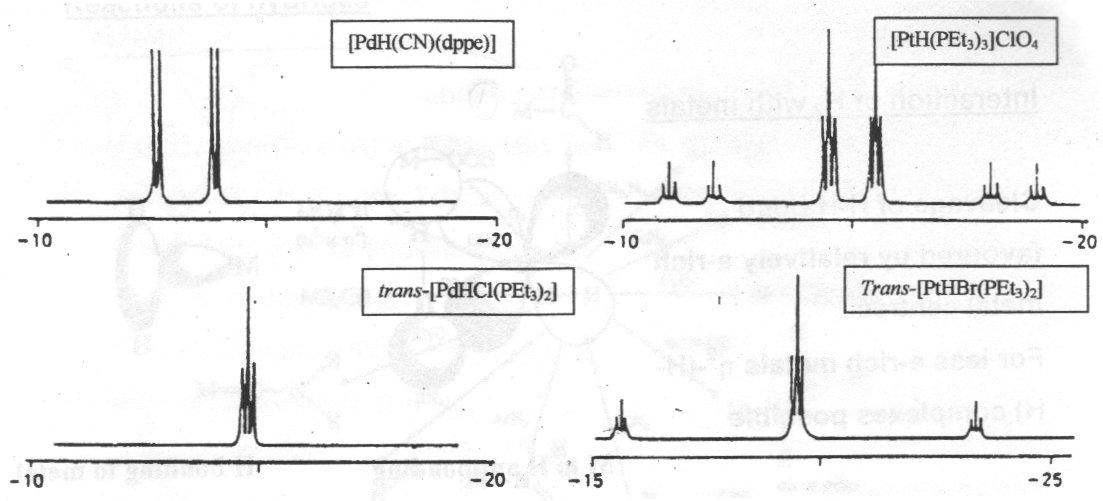
Note Pt spectra show expected satellites due to coupling to 1/3 195Pt.

[ReH9]2- -
Prepared by reaction of [ReO4]- with Na metal in EtOH.
Structure by neutron diffraction.
Tricapped trigonal prism:
[W(H2)(CO)3(Pchex3)2] –
Synthesis –
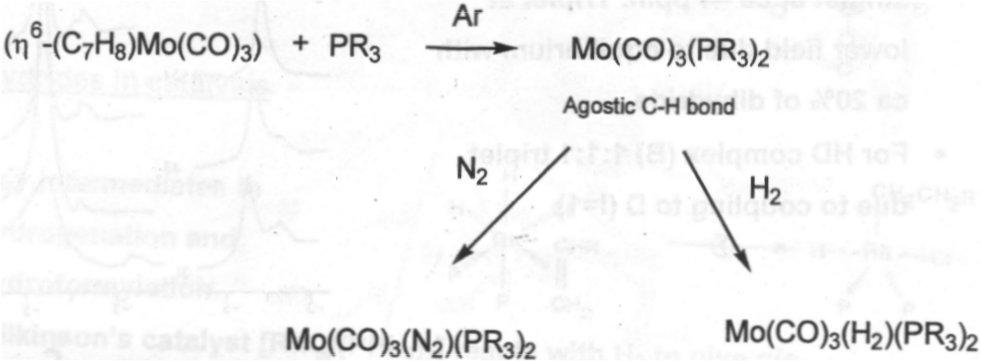

- Reaction under Argon gives apparently 5 coordinate species, but agnostic interaction occupies 6th site. Reaction with H2 gives dihydrogen complex.
- Neutron diffraction structure shows H-H bond orthogonal to W(CO)3 plane.
- Some disorder in the iso-propyl groups of the phosphine.
- IR Spectroscopy shows H-D at 2360cm-1.
- 1H NMR for H2 complex at room temperature has broad singlet at about -4ppm. Triplet at lower field due to equilibrium with 20% of dihydride.
- For HD complex, 1:1:1 triplet due to coupling to D (I=1).
Many other dihydrogen complexes now identified – often made by protonation of hydrides.
Basicity of Hydride Complexes –
pKa’s of hydrides dependent on M-H strength and nature of co-ligands: [HCo(CO)4] is a strong acid, while [HCo(CO)3(PPh3)] is neutral.
Many hydrides particularly to the right of the transition metals have electron rich metals and protonation occurs at the metal.
Bridging Hydrides –
Usually supported by M-M bonds, common feature of metal carbonyl clusters. A few have no M-M bond e.g. [HW2(CO)10]-. Evidence for bridging from Neutron or X-ray Diffraction, and from NMR where coupling to two metals can be seen for metals.
Also possible to have H encapsulated inside octahedral cluster, e.g. [Co6H(CO)15]-. 1H NMR shows hydride at +23.3ppm.
Hydrides in Catalysis –
Key intermediates in hydrogenation and hydroformylation.
Wilkinson’s Catalyst [RhCl(PPh3)3] reacts with H2 to give cis-[RhH2Cl(PPh3)3] and loss of PPh3 then provides site for olefin coordination. Two subsequent insertion reactions into M-H liberate the hydrocarbon.
Reactions of Hydrides –

The Post-Transition Metals
|
Group 11 |
Group 12 |
Group 13 |
Group 14 |
|
Cu |
Zn |
(Ga) |
(Ge) |
|
Ag |
Cd |
In |
Sn |
|
Zn |
Hg |
Tl |
Pb |
Ionisation Energies –

Figures above show plots for I1, I1+I2, I1+I2+I3, and I1+I2+I3+I4.
- I1 values for Cu, Ag, and Au higher than Group 1 due to increase in Zeff due to poor shielding by nd10 cores, and increasing penetration of s.
- IE’s for Group 13 lower as p electrons being removed.
- Alternation effect for rows 3-5.
- For 3rd row, increase in I1+I2+I3 due to poor shielding of 3d10 – increase in Zeff.
- For 4th row increased number of electrons outweighs increase in Zeff.
- For 5th row poorly shielding 4f intervene, and relativistic effects – increase in IE.
- Values for formation of M3+ and M4+ higher than for equivalently charged TM ions.
Heats of Sublimation –
- Values for Group 11 higher than Group 1, as ns,np and (n-1)d all involved in metallic binding.
- Group 12 much lower as d orbitals move into core. Decrease as orbitals contract due to increase in Zeff. Low value for Hg accounts for liquid character.
- Values generally 2-300kJmol-1, much lower than Transition Metals. Small compared to IE’s discussed above.
Ionic Radii –
- Large increase in Zeff causes large contraction in size for Cu+ cf. Li+.
- Similar effects for 4th and 5th rows Ag+ cf. Na+.
- Less marked for subsequent groups Tl+ cf. Rb+.
- Zn2+, Cd2+, and Hg2+ all smaller than comparable Group 2.
- Heats of hydration follow trends in ionic size with Ag+ having comparable value to Na+.
Redox Potentials –
- Values dominated by high IE term.
- Group 11 metals all electropositive, do not react with acid to give H2, readily easily reduced to metal, poor reducing agents.
Inert Pair Effect
There is much data that suggests that the (n-2) oxidation state for Groups 13, 14 and 15 increases in stability relative n Oxidation State with descent of the groups.
Decomposition MCl4 → MCl2 + Cl2 increasingly thermodynamically favoured down Group 14. Also lattice energy of MCl2 also stabilises M(II) state.
Redox potentials indicate that Pb(IV) and Tl(III) relatively oxidising.
Rationale –
- For ns2npx elements formation of MX4 species requires promotion of s electrons to p to form necessary hybrids. Promotion energies s to p follow similar trend to IE’s.
- Strength of M-X bonds decreasing with descent of group, less energy available to compensate for promotional energy expenditure. Effect is stabilisation of M(II) wrt M(IV) and M(I) wrt M(III).
Lone pair of electrons can have stereochemical consequences, thus SnCl2 has a bent structure with a lone pair, cf. ZnCl2 which is linear.
Metal-Metal Bonding
In Group 13, GaCl2 is actually Ga+[GaCl4]-, but addition of halide ions gives [Ga2Cl6]2-, which has a Ga-Ga bond with staggered terminal Cl groups. Subtle balance between M(II) and M(I)/M(III) – disproportionation. Similarly Pb3O4 contains Pb(II) and Pb(IV).
For Hg disproportionation, Hg22+ → Hg2+ + Hg has K = 1.14x10-2, so a ligand that precipitates out Hg2+ e.g. sulphide will drive equilibrium to the right. Hg-Hg bond 250-270pm. Species Hg32+ and Hg42+ also known by AsF5 oxidation of Hg metal giving [AsF6]- anion. Zn2+ and Cd2+ not stable in aqueous solution.
Zintyl Cations and Anions
So called naked clusters can be prepared:
BiCl3 + Bi + NaAlCl4(melt) → [Bi5]3+[AlCl4]3
NaSn1.7 + crypt → [Na(crypt)]2[Sn5]2-
Reactions carried out in non-aqueous solvents to prevent hydrolysis.
E5 species are isoelectronic, and structure can be interpreted in terms of Wades Rules.
Stabilities of Complexes –
Plots are of stepwise formation constants for formation of NH3 and halide complexes:
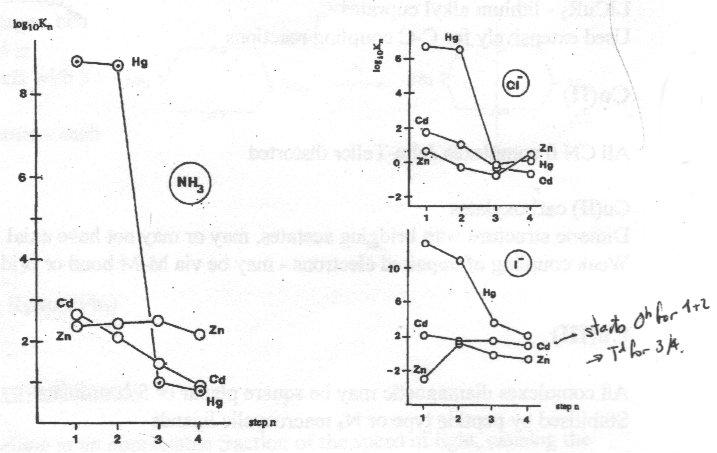
Left-hand plot for NH3 complexes shows discontinuity for Hg for addition of 3rd NH3 – reflects preference for 2 coordination.
Right-hand plot for halides shows preference of Hg and Cd for softer iodide.
K values for Zn indicate that it is a class A, hard acceptor.
Scale of Y axis masks effect of change in coordination number for Cd/I- complexes (earlier).
Two Coordination –
Data above for Hg illustrates tendency for two coordination increases down the group.
Attributed to large s-p energy difference for heavier elements which disfavours formation of hybrids for higher coordination numbers.
For Cu, Ag, Au also possibility of use of dz2 orbitals to form d-s hybrid – ideal for 2 coordination.
Reluctance for coordination of anionic ligands (+1 charge).
Brief Notes on Descriptive Chemistry of Group 11
Copper –
Cu(I):
- Common geometries – linear: [CuCl2]-, trigonal: [BrCu(μ-Br)2CuBr], tetrahedral: [Cu(MeCN)4]+
- Complexes generally colourless.
- Cu(II)/Cu(I) equilibrium strongly dependent on co-ligands. Addition of I- favours Cu(I), en Cu(II) via [Cu(en)22+
Tetrameric structures common:
e.g. [CuXL]4 where X = halogen, L = py, PR3.
Can have cubane or step structure.
Ag(I) analogues isostructural.
Cu(I) organometallics:
LiCuR2 – lithium alkyl cuprates. Used extensively for C-C coupling reactions.
Cu(II):
All coordination 6 complexes are Jahn-Teller distorted.
Cu(II) carboxylates – dimeric structure with bridging acetates, may or may not have axial ligands. Weak coupling of unpaired electrons – may be via M-M bond or bridging acetates.
Cu(III):
All complexes diamagnetic, and may be square planar or 5 coordinate.
Stabilised by peptide type or N4 macrocyclic ligands.
Silver –
Ag(I):
Common geometries – linear: [Ag(NH3)2]+, Tetrahedral: [Ag(PPh3)4]+, Octahedral: AgCl (NaCl structure).
Typical class b soft metal ion, good affinity for P, S donors, poor for O.
But [Ag(S2O3)]- known – AgBr dissolves in thiosulphate.
Ag(SR)n cage molecules known (no M-M bonds) – value of n depends on steric requirements of R.
Ag(I) oxide, Ag2O – brown ppt from addition of OH- to Ag(I), strongly basic due to formation of AgOH.
Ag(II):
Ag2+/Ag couple +2V in acid – powerful oxidant.
Can be stabilised by N-donors, e.g. [Ag(py)4]2+.
Paramagnetic, square planar structure.
Ag(III):
By anodic oxidation of Ag in alkaline solution.
Can be stabilised with polydentate N ligands, and periodate.
Gold –
Au(I):
2 coordination dominates, e.g. [AuCl2]-, with strong class b donor ligands higher coordination numbers are found, e.g. [AuCl(PPh3)3].
Complexes of type {Au(SR)}n have been used extensively to treat arthritis – exact mechanism still uncertain.
Wide range of metal cluster complexes of type, e.g. [Au13Cl2(PMe2Ph)10]3+ available by controlled reduction of [AuCl(PPh3)] with borohydride. No Ag equivalents – reflects increased tendency for M-M bonding.
Au(II):
Difficult to stop oxidation of Au(I) at Au(II) stage – but can be stabilised by planar N4 macrocycles and in binuclear complexes bridged by didentate ligands with 3 atoms in the bridge.
Au(III):
Mostly square planar (d8), [AuCl4]-, [Au(NH3)4]3+.
Range of organometallic derivatives known, e.g. R3Au(PPh3).
Relativistic Effects for Au and other 5th Row Elements
For the heavier elements the electrons are travelling at an appreciable fraction of the speed of light, causing the mass to increase and thus the orbital size to decrease, and energetic stabilisation of the orbital.
Electrons accelerate close to the nucleus, and therefore effect largest for most penetrating, and s orbitals are contracted most.
d orbitals penetrate little, and are not therefore contracted, in fact the contraction of the s, and to lesser extent p orbitals, increases their shielding ability, and in fact therefore the d and f orbitals undergo an indirect relativistic expansion.
In the case of Au it appears that a large contribution to the contraction of the 6s orbital is from the expansion (and therefore poorer shielding) of the inner 5d and 4f orbitals, effect is largest at Au.
It has been argued that the M-M bonding found in certain types of Au)I) complexes is due to 6s/5d hybridisation, facilitated by relativistic effects, but still subject od debate.
For 3rd row transition elements the relativistic expansion of the 5d orbitals and destabilisation means they can overlap better with π-acceptor ligands such as CO – calculations suggest contribution of about 20kJ mol-1 to M-CO bond strength (i.e. about 10%).
The Lanthanides and Actinides
The Lanthanides
|
|
La |
Ce |
Pr |
Nd |
Pm |
Sm |
Eu |
Gd |
Tb |
Dy |
Ho |
Er |
Tm |
Y |
Lu |
|
M3+ |
4f0 |
4f1 |
4f2 |
4f3 |
4f4 |
4f5 |
4f6 |
4f7 |
4f8 |
4f9 |
4f10 |
4f11 |
4f12 |
4f13 |
4f14 |
Thermodynamic trends

Plot shows trends in – A = I3, B = I1 + I2 + I3, C = heats of hydration
- Shape of plots for A and B reflects the fact that exchange energy is at a maximum for the half-filled shell (for M(II) this is at Eu), thus IE highest at this point.
- Discussed in some detail for TM case
- I4 will follow directly similar pattern to I3, displaced one element to right
- Also smaller inflections at ¼ and ¾ filled shells, e-e repulsion function of angular momentum.
- Heats of hydration C smooth decrease across series – follows ionic size, which decreases gradually across series
Heats of atomisation
Much smaller the IE’s and trends mirror those of IE as metals have configurations 4fn5d16s2 whereas atoms are 4fn+16s2.
I3 trends dominant.
Consequences for chemistry
Stability of M(II) state:- high values of I3 at Sm and Eu mean Eu(II) and Sm(II) stable
Stability of M(IV) state: low values of I4 at Ce and Pr and Tb mean that M(IV) states stable for these elements.
Magnetic properties and spectra
- The electrons of 4f orbitals do not quench the orbital angular momentum as in the case of TM’s, and states of lanthanide complexes essentially those of the free ions –
- Determined by Russell-Saunders coupling scheme.
- Spin-orbit coupling is large, and no excited states are thermally accessible.
- Magnetic moment given by expression:

- For Nd3+ ground state 4I9/2, hence magnetic moment calculated as 3.62, in good agreement with experimental value of 3.5-3.6.
- Fits well for all Ln3+ except Sm(III) and Eu(III). Here there are low-lying excited states that are thermally accessible, and the magnetic moments are temperature dependent.
- f-f transitions weak as Laporte forbidden.
- No crystal field effects, and lines very sharp, as no broadening M-L variations.
Coordination chemistry
For aquo ions, CN’s generally 8,9. For 9 coordination geometry capped trigonal prism.
As expected CN’s decrease across series with decreasing ionic size (10% reduction across series)
Some hydrolysis in neutral solution.
Solubilities of Ln3+ depends on delicate balance lattice and salvation energies/entropy.
In water stable complexes only formed with polydentate, charged I donors e.g. [Ln(acac)3(H2O)]
Can be dehydrated to give [Ln(acac)3]. More lipophilic acac derivative gives complex used as shift reagent in NMR – presence of paramagnetic centre causes shifts in proton NMR peaks
Use of non-aqueous solvents permits isolation of complexes with N donors e.g. [Ln(en)4]3+.
Use of bulky amide ligands permits isolation of 3 coordinate complexes [Ln{(Me3Si)2N}3] – planar in solution but pyramidal in solid state
M(II) State
- Prepared by electrolysis of Ln3+ - Sm2+ dark red, Eu2+ colourless, Yb2+ yellow.
- Readily oxidised by air and Sm2+ reduces water.
- Eu and Yb dissolve in liquidNH3 to give blue solutions containing [Ln(NH3)2]2+ and solvated electron (cf group 2)
Organometallic chemistry

No CO chemistry as f-orbitals cannot participate in synergic bonding
Dominated by cyclopentadienyl chemistry – primarily ionic bonding. Very reactive to air and water.
Synthesis: MCl3 + nNaCp

Use of pentamethyl Cp prevents binding 3rd Cp.
(Me5C5)2LuMe has range of novel activity.
Catalyses olefin polymerisation
Also activates saturated hydrocarbons – cf Bergmann Cp*Ir(PMe3) system

Also Cp chemistry of Ln(II) state
Side on briging mode for N2 molecule, similar to that found for Zr complexes
Also some chemistry of cyclooctatraene (COT)
CeCl3 reacted with K2[COT] in diglyme to give [K(diglyme)][Ce(COT)2]
COT rings are staggered, and one also bound to K.
Many applications for Ln compounds, phosphors, replacement for Ca2+ in metalloenzymes, lasers (Neodymium YAG laser), LaNi5 for hydrogen storage.
Comparison of lanthanides and transition metals
|
Property |
Lanthanide Ions |
1st row transition metals |
|
Metal orbitals |
4f |
3d |
|
Ionic radii |
106-85pm |
75-60pm |
|
CN’s |
6-9 |
4-6 |
|
Geometries |
Trigonal prismatic, square antiprismatic, capped tbp, dodecahedral |
Square planar, tetrahedral, octahedral |
|
Bonding type |
Little interaction of L with 4f orbitals |
Strong ligand 3-d interactions |
|
Direction of bonds |
Little preference |
Strongly directed |
|
Bond strengths |
Correlate with electronegativity F>OH>H2O>Cl- |
Spectrochemical series CN>H2O>F- etc |
|
Type of complexes |
Ionic, rapid ligand exchange |
Considerable covalency, ligand exchange dependent on no d electrons |
The Actinides
|
|
Ac |
Th |
Pa |
U |
Np |
Pu |
Am |
Am |
Bk |
Cf |
Es |
Fm |
Md |
No |
Lr |
|
Z |
89 |
90 |
91 |
92 |
93 |
94 |
95 |
96 |
97 |
98 |
99 |
100 |
101 |
102 |
103 |
|
M2+ |
7s |
5f6d |
5f26d |
5f36d |
5f5 |
5f6 |
5f7 |
5f8 |
5f9 |
5f10 |
5f11 |
5f12 |
5f13 |
5f14 |
5f147s |
|
M3+ |
|
5f |
5f2 |
5f3 |
5f4 |
5f5 |
5f6 |
5f7 |
5f8 |
5f9 |
5f10 |
5f11 |
5f12 |
5f13 |
5f14 |
- All isotopes of all elements are radioactive. Heavier elements undergo alpha decay – particularly dangerous if ingested.
- Only Ac, Th, Pa and U occur naturally.
- Heavier elements made by neutron or heavy ion bombardment of lighter elements – only isolated a few atoms at a time.
- 238Pu gives out heat due to alpha decay (to 234Pu) – use as heat source.
General Trends
- 5f, 6d, 7s, 7p orbitals all of comparable energy – electronic structures difficult to assess.
- Ionic radii decrease across series.
- Electronic spectra sharp, more intense than Ln.
- Magnetic moments tough to interpret.
- M(III) most common state for all An, particularly post Am.
- M(IV) important for Th, Pa, U.
- M(V) most stable for Pa.
- M(VI) in form AnO22+ for U, Np, Pu.
- Coordination Numbers range from 4 to 14.
Chemistry of Uranium
Oxides
- UO3 – by heating uranyl nitrate at about 500oC, structure octahedral – UO2 + 4 other atoms.
- U3O8 – by heating uranyl nitrate at about 700oC, contains planar UO7 units.
- UO2 – reduce UO3 with H2 – fluorite structure, extra interstitial oxide anions can be added to give U4O9.
Halides
- UF6 – from UF4 + ClF3 – octahedral, used on large scale to separate 235U from 238U by gaseous diffusion.
- UCl6 – U3O8 + C + Cl2 – decomposes in solution to give U2Cl10 – octahedral structures with bridging halide.
- UCl4 – UO3 refluxed in hexachloropropene, forms 9-coordinate adducts.
- UCl3 – from UH3 + HCl, chemistry similar to LnCl3.
Range of halogeno complexes known for U(III) to U(VI).
Aqueous Chemistry
Frost diagram shows:
- U(III) reduces water.
- U(IV) stable, but aquo ion hydrolysed at neutral pH.
- UO2+ unstable wrt U(IV) and U(VI).
UO22+ - uranyl ion – has trans arrangement of oxo-groups due to participation of both d and empty f orbitals. U-O bonds best viewed as involving 1 σ and 2 π bonds.
Readily bonds ligands in equatorial plane (e.g. 2NO3, 2H2O).
Organometallic Chemistry
Like Ln elements, Cp chemistry is important.
Three main types:
Cp4An, Cp3An and Cp3AnX (X = Cl, OR, BH4, alkyl).
Prepared from appropriate halide and NaCp.
Green uranocene, [U(COT)2] from UCl4 + 2K2[COT]. Eclipsed arrangement of rings, paramagnetic with 2 unpaired electrons.
I am happy for them to be reproduced, but please include credit to this website if you're putting them somewhere public please!