Rearrangements
Some basic notes at the start, then covers examples and mechanisms later on.
Rearrangements Notes
Mechanistic Aspects of Rearrangements
Nature of the Rearrangement
It can vary from being truly stepwise to migration occurring in concert with initial ionisation. These two situations can be considered as intramolecular analogues of SN1 and SN2 respectively. Hence, it has profound stereochemical implications at both the migration origin and the terminus if the centres are chiral. With non-racemic substrates, the generation of a free carbocation at either the migration terminus or origin will result in loss of stereochemical integrity at that centre, whereas a concerted migration with no intermediate will result in inversion of the absolute stereochemistry at the terminus.
Migratory Aptitude
This is the ease with which any particular group will undergo nucleophilic 1,2-shifts. It is related to electron donating capacity, but such values are not quantifiable as a single group will show different aptitudes for different reactions/conditions.
Note that in some instances, for example the Beckmann Rearrangement, only one group ever migrates regardless of the aptitude, simply because of stereoelectronic requirements for the reaction. Likewise, in for example the pinacol rearrangement, the most stable cation controls the reaction pathway, as opposed to the migrating group.
A wide range of other factors, such as steric and conformational effects, play subtle roles in determining whether a particular migration is favoured. Generally however, aryl groups migrate more readily than alkyl groups, and within the aryl series, e-donating substituents increase the propensity for migration. However, the position of hydrogen within this framework is highly unpredictable.
Neighbouring Group Participation (Anchimeric Assistance)
This is where a substituent is capable of stabilising an adjacent carbocation by acting as an intramolecular nucleophile. More details in the Stereoelectronics Notes.
Non-classical Carbonium Ions
While alkyl groups do not usually undergo σ-participation in acyclic or unstrained ring systems, there is much evidence to suggest that this does occur in strained rings. The species formed contain a two-electron, three-centre bond and are known as “non-classical carbonium ions”. The most studied system in which σ-participation is envisaged to occur is the 2-norbornyl system. It has been found that solvolysis of optically pure norbornyl 2-exo-brosylate in acetic acid gave a racemic exo-acetate as the sole product with no endo-material. Furthermore, it occurred 250 times faster.
These results were interpreted as implying that the reaction of the exo-substrate
occurred solely via a non-classical carbonium ion, whilst the endo substrate reacted by initial
formation of
a classical carbenium ion which then rearranged to the non-classical carbonium ion, but not before a
small
amount had reacted with solvent, attack being sterically directed to the exo-face.

Carbocation Induced Alkyl and Hydride Shifts
Wagner-Meerwein Shifts
These 1,2-alkyl shifts occur if it leads to the new carbocation being more stabilised than the original. This is usually the case if the carbon adjacent to the positively charged centre is secondary or tertiary. Subsequent elimination will then occur. Saytzeff’s Rule is obeyed to give the most substituted alkene.
1,2-alkyl shifts have a rigorous stereochemical requirement for the bond at the migration origin to lie very close to the axis of the vacant p-orbital of the carbocationic centre. Whilst acyclic systems can usually adopt the correct conformation for this alignment, this option may not be open to cyclic systems.

Pinacol Rearrangement
Treatment of 1,2 diols with acid, which converts them to carbonyls. The reaction can also occur on β-aminoalcohols, halohydrins, epoxides and allylic alcohols.
It proceeds via a 1,2 alkyl shift, and the overall reaction is:

Control can be exerted by conditions, e.g.

Tiffeneau-Demjanov Reaction
Similar to the pinacol rearrangement, but via a β-amino alcohol derivative. This specific reaction leads to one carbon ring expansion, and is very useful for homologating cyclic ketones to give products containing 4-8 carbon atoms in the ring.


This type of reaction can also occur with other suitable leaving groups, e.g. bromohydrins.
Dienone-Phenol Rearrangement
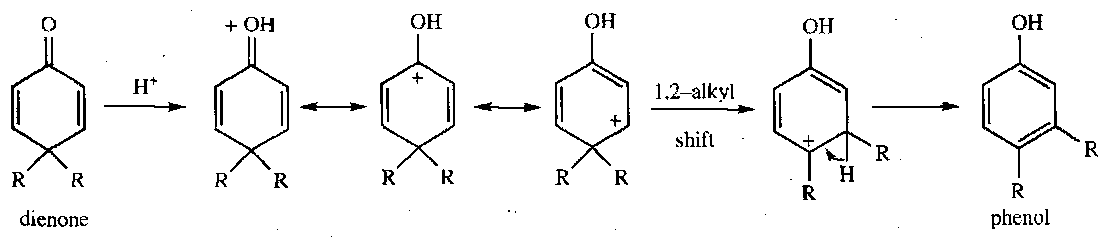
Hydride Shifts
Less common than alkyl shifts, as the latter usually confers steric relief. 1,2-H shifts are typically seen where steric requirements override this control.
Particular examples include transannular hydride migrations, where H’s move over large distances in medium (C8-11) rings. This is possible as with these rings the H’s can actually be quite close to other parts of the ring, as shown:
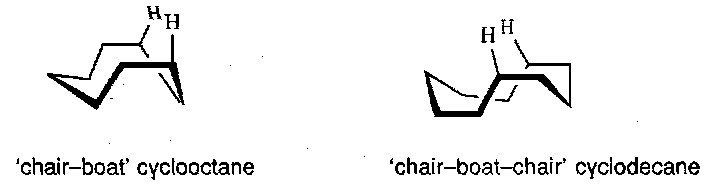
Such shifts were first noted when the acid catalysed opening of medium ring epoxides was found to give only small amounts of the desired trans-1,2-diols.

Nucleophilic Rearrangements to Carbon (involving carbenes and carbanions)
Favorskii Rearrangement
Rearrangement of α-haloketones. Particularly useful for ring contraction. Labelling was useful in showing the mechanism:

Ramberg-Backlund Rearrangement
Another rearrangement via a cyclic intermediate, and refers to the base promoted conversion of α-halosulphones into alkenes.

Benzil-Benzilic Acid
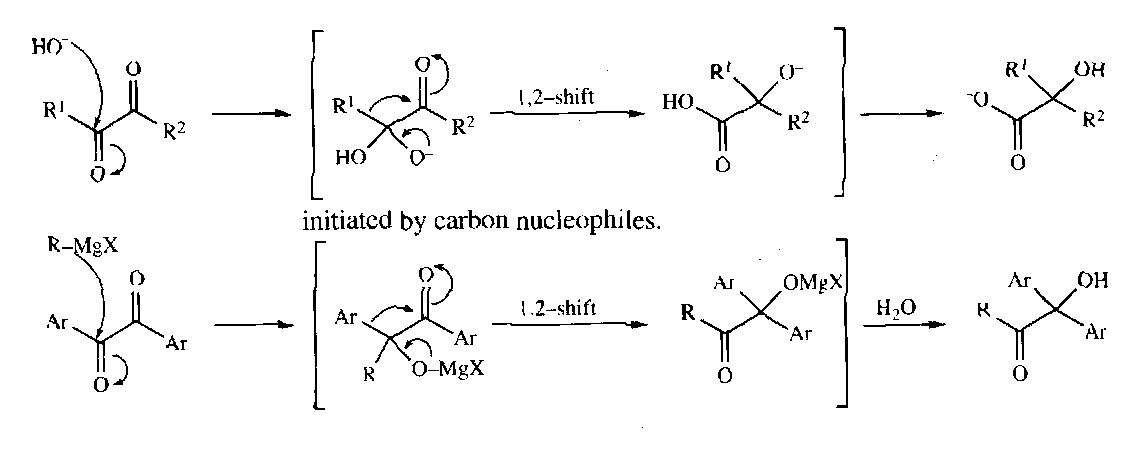
Wolff Rearrangement

The ketene is usually attacked by an alcohol to give an ester.
Arndt-Eistert Reaction
Modification of Wolff Rearrangement.
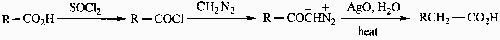
Rearrangements to e-deficient N
Beckmann
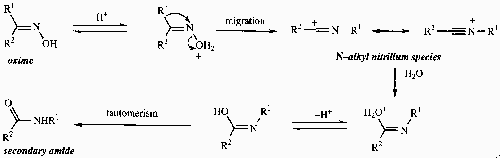
The most important point to remember here is that the R group anti to the oxime N-O bond is the one that migrates (i.e. R1 in the above), regardless of migratory aptitudes.
With certain substrates, particularly those in which one of the substituents on the oxime is a tertiary alkyl group or possess a β-heteroatom substituent, a nitrile may be formed by a fragmentation process instead of a rearrangement. This is the Secondary Beckmann Reaction, and is a consequence of the enhanced stability of the carbocation produced under these circumstances.

Neber Rearrangement
Similar reagents to the Beckmann Rearrangement, except for the important distinction that the Neber occurs in base, while the Beckmann is in acid. The key synthetic difference ins that there is no dependence on stereochemistry of the oxime sulphonate, i.e. which substituent possesses the most acid α-position migrates, not necessarily the anti one.

Curtius Rearrangement
Carboxylic acid to primary amine with one less carbon atom. Prepare isocyanate:

Hydrolysis:

Lossen Rearrangement
Mechanistically very similar to Curtius Rearrangement. Difficult to obtain starting material though, although only requires water to carry out. Base catalysed.

Schmidt Rearrangement
Similar steps to Curtius above, although formation of acyl azide differs, being acid catalysed:

Hofmann Rearrangement
Similar net result to the above 3 rearrangements, but the formation of the isocyanate again differs.

Stieglitz Rearrangement
Nucleophilic migration from carbon to nitrogen.

Rearrangements to e-deficient O
Baeyer-Villiger Rearrangement
This is a rearrangement to electron deficient oxygen. Converts ketones to esters, and cyclic ketones to lactones.

Dakin Reaction
Aromatic aldehydes and ketones possessing a hydroxyl or amino group in either the ortho or para position can be converted to phenols on treatment with alkaline hydrogen peroxide. Mechanistically similar to Baeyer-Villiger.

Electrophilic Rearrangements
Stevens

Sommelet-Hauser Rearrangement

Meisenheimer Rearrangement
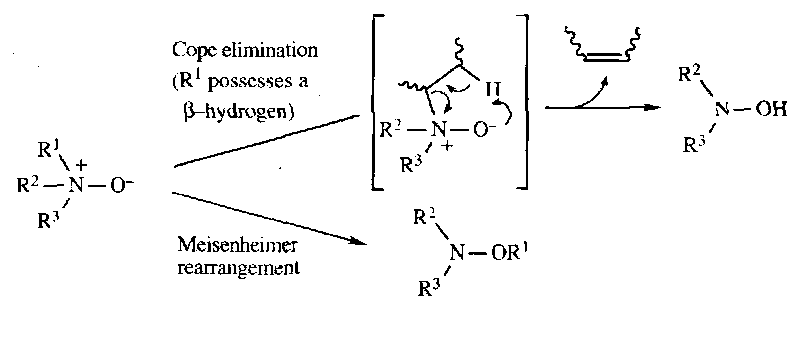
Similar to the two preceding reactions. Tertiary N-Oxide undergoes thermal rearrangement, where the migrating group is usually benzylic or allylic. If any of the groups possess a β-hydrogen, Cope elimination predominates, limiting the synthetic utility of this rearrangement.
Wittig Rearrangement
Analogous to the Stevens Rearrangement above, but Oxygen instead of Nitrogen. It also requires stronger bases than Stevens.

Smiles Rearrangement


Carbon to Carbon Electrophilic Migration
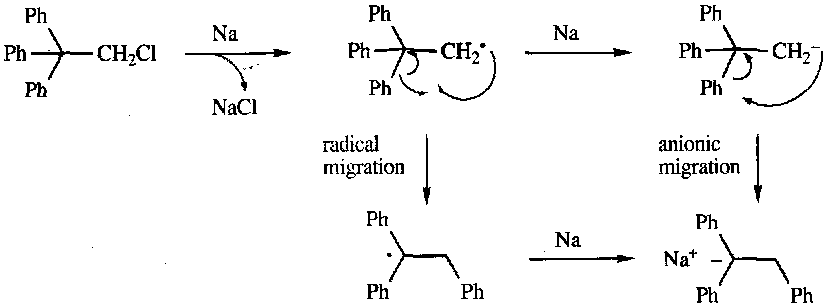
Note that these are far rarer than the carbocation induced nucleophilic migrations encountered earlier. Requires exceptional circumstances of steric and electronic effects in combination.
It is known to occur in metallation of triaryl-2-haloalkanes, e.g.
Acid Catalysed Aromatic Rearrangements
Jacobsen Rearrangement

Note that the product above can be desulphonated (by reversing the first step), such that the Me groups have been rearranged. The reaction is driven to the point where the most stable arenium ion is formed.
Fries Rearrangement
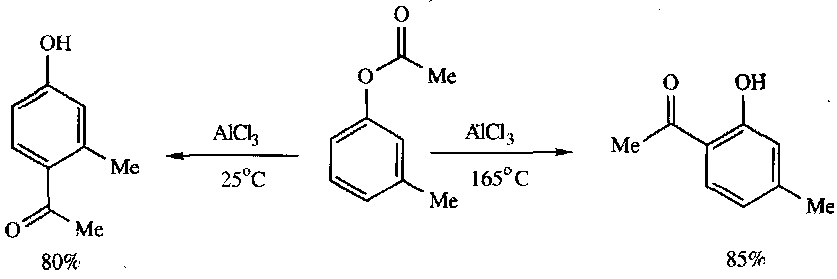
Note the temperature control of the products. The mechanism is not well understood with regards to whether it is inter or intramolecular migration.
Note that there are similar reactions which involve the migration of the R group from an aryl ether, and similarly the R group from an alkylarylamine. Both lack stereochemical control though.
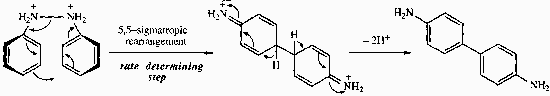
Benzidine Rearrangement

The mechanism for this is:
I am happy for them to be reproduced, but please include credit to this website if you're putting them somewhere public please!