Protecting Groups and Carbohydrates
Covers Protecting Groups (On and Off methods, although omitted mechanisms as they're very easy, and then moves onto some useful sugar transformations (although this doesn't often come up on Finals any more)
Protecting Groups & Carbohydrates Notes
Protecting Groups
Protecting Groups are introduced onto a Functional Group to block its reactivity under experimental conditions needed to make modifications elsewhere. The negative aspect of this is that taking them on/off results in a loss of yield.
Criteria for a good protecting group:
- Quantitative yield at desired site.
- No alteration to the rest of the molecule.
- Preferably no new stereogenic centres.
- Not broken down in subsequent stages.
- Does not cause side reactions.
- Removable under specific conditions.
Also things like cost / toxicity etc.
Reaction Conditions should be available that remove one type of protecting group, leaving others intact. This is an Orthogonal Strategy. We may require a degree of fine-tuning to allow selectivity (graduation of lability) so this is important. A good example is the effect of silyl protection bulk on ease of removal.
Ethers / Alcohols
Benzyl
ON = base then Benzyl Chloride.
OFF = hydrogenation (Pt/H2).
Paramethoxybenzyl (PMB)
ON = same as benzyl, except use (p-OMe)Benzyl Chloride.
OFF = DDQ or CAN (NH4)2Ce(NO3)6, i.e. one electron donation (SET).
Silyl
ON = silyl chloride, base (useful to soak up base). Often a nucleophilic catalyst is needed for bulkier silyls, such as imidazole.
OFF = F- or acid, via Ates Complex. Based on strong Si-O/Si-F bonds.
Selectivity – 1o > 2o > 3o alcohols, i.e. based on sterics (accessibility and lability).
Specific examples are:
TMS – Me3Si – easy to add/remove, but sometimes too easy. Vulnerable to oxidation conditions. Good for temporary protection.
TES – Et3Si – useful in that easier to remove relative to other bulkier Si protecting groups → selectivity. Used particularly in OH → =O oxidation (CrO3, Swern), as these conditions remove TES but not bulkier silyls.
TPS – Ph3Si – very easy to remove in basic conditions but slowed in acid (between TES and TBS, see below). Also very bulky → diastereocontrol, although less good at this than TBS.
TBS – tBuMe2Si – 1o > 2o, but doesn’t protect 3o alcohols unless TBSOTf + pyridine is used. It is cleaved by AcOH, ArSO3H and F-.
TBDPS – tBuPh2Si – more extreme versions of TBS. Very tolerant to acid conditions, but not basic.
TIPS – iPr3Si – the most stable to base/nucleophilic conditions.
Silyls will also protect aldehydes / ketones by trapping the enol ether. Si-N bond is weak, so this does not work for amines.
Allyl
ON = allylic bromide + base.
OFF = HgX2 + H2O/H+ (hydrolysis). The C=C bond complexes to the Hg. Strong base can also be used instead (tBuOK), or oxidative cleavage (OsO4).
Trityl
ON = Ph3C-Cl. Attaches to primary alcohols only. SN1 mechanism.
OFF = Weak acid. It resists base, but is easily removed in acid due to the stability of the carbocation. HCO2H will cleave it in the presence of other acid-sensitive protecting groups.
Acetals
Acetonide
ON = acetone/H+(cat), Me2C(OMe)2.
OFF = aqueous H+.
See Carbohydrates Notes for the mechanisms for this.
Benzylidene
ON = PhCHO / H+(cat), or PhCH(OMe)2.
OFF = aqueous H+, or hydrogenation, or reductive opening.
See Carbohydrates Notes for the mechanisms.
Tetrahydropyran (THP)
ON =
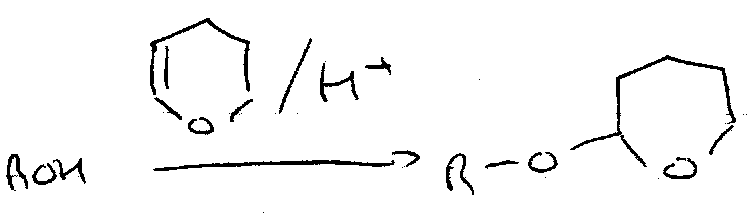
OFF = Aqueous acid (Acetal Hydrolysis).
Methoxymethyl Ester (MOM)
ON = MeOCH2Cl + NiPr3. Adds to 1o and 2o only. 3o requires MeOCH2I. These reagents are highly toxic, so there are some variants. Borderline SN1/SN2 mechanism.
OFF = strong H+. Can be a problem for the rest of the molecule. Lewis Acid / Nucleophilic combinations are more selective.
2-methoxyethoxy methyl, MEM
Lewis Acid labile. More SN1 character. Methyl ethers are good due to reluctance to form Me+.
2-trimethylsilylethoxymethyl, SEM
Me3SiCH2CH2OCH2X. Protect similar to other acetals, but deprotection is orthogonal as it is the same as silyl ethers (i.e. H+ or F-).
Esters
Acetate
ON = Ac2O or AcOCl + base (pyridine usually).
OFF = base (-OMe).
Benzoyl
ON = PhCOCl.
OFF = Base (-OMe).
Pivaloate
ON = Me3COCl.
OFF = Base (-OMe).
Carboxyls
Orthoester
This is the acetal of an ester, i.e. RC(OR’)3. Inefficient forming from acid, but good for cyanide.
They are more acid labile than acetal/R2CO or equivalent, due to extra stability of (MeO)2C+.
Trichloroethoxycarbonyl, Troc
This is ROCH2OCH2CCl3. The –OCH2CCl3 fragment is labile to reduction, so removal is usually achieved by Zn with H-solvent. This is advantageous as Zn reacts slowly with most functional groups (other than halides), so highly selective deprotection.
N-protection
Carbamates
R2NCO2R’ group. Amine + alky chloroformate in aq. NaOH. They work by deactivating the Nitrogen nucleophilicity, and the carbonyl is stable to other nucleophiles (amide and ester).
O-alkyl groups are chosen to allow mild and specific deprotecting, as in Peptide Synthesis.
Troc
See above.
Fmoc
This is fluorenylmethyloxycarbonyl. See Peptides Notes.
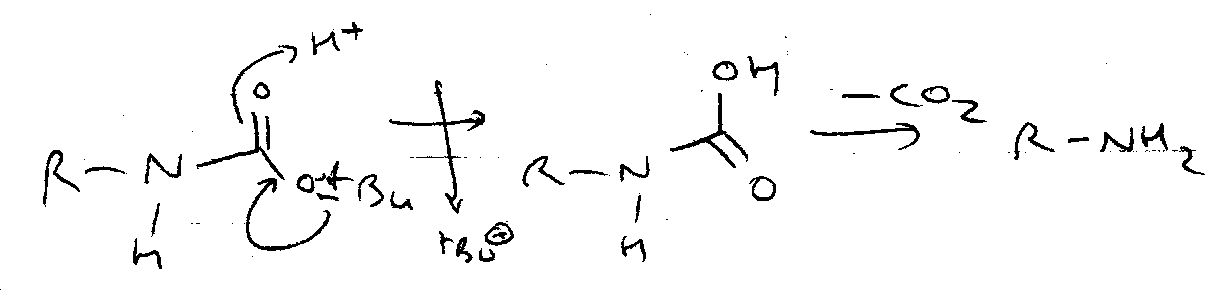
Boc
ON =

OFF =
Alloc
ON =
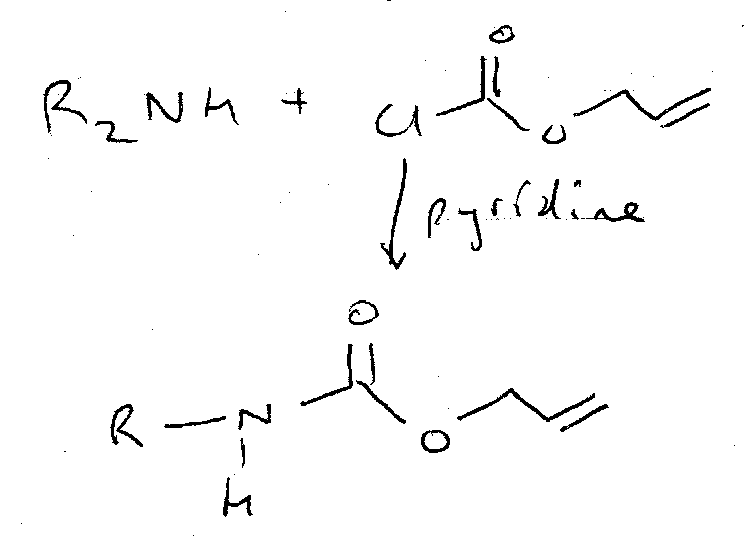
OFF =
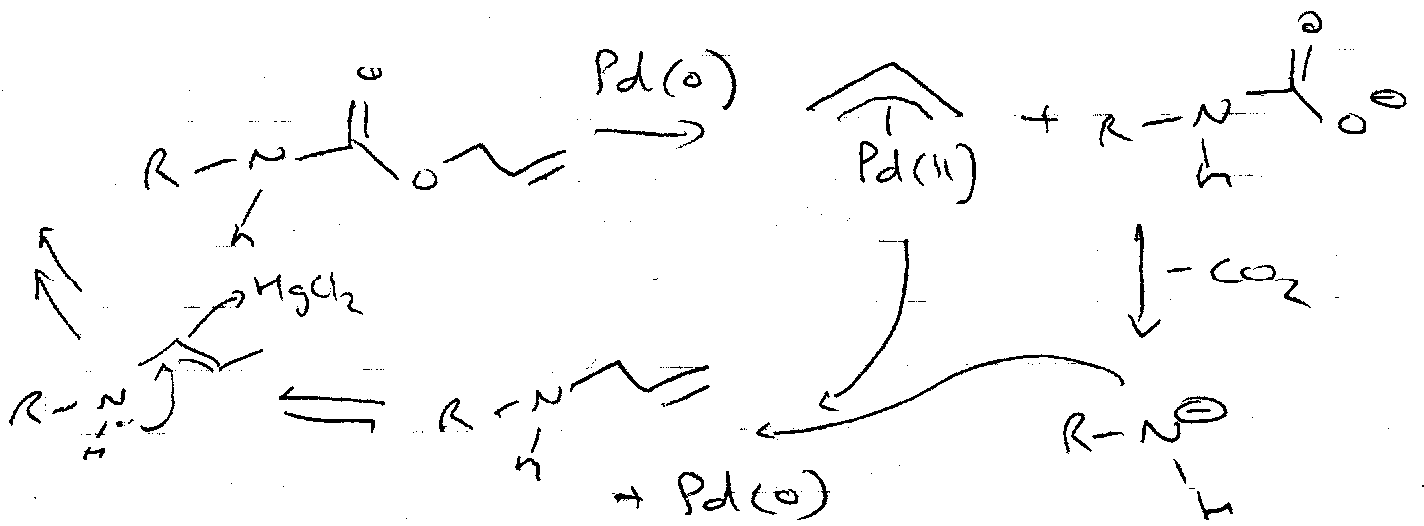
CBz
PhCH2OCOX. This is acid labile (not as much so as Boc).
Carbohydrates
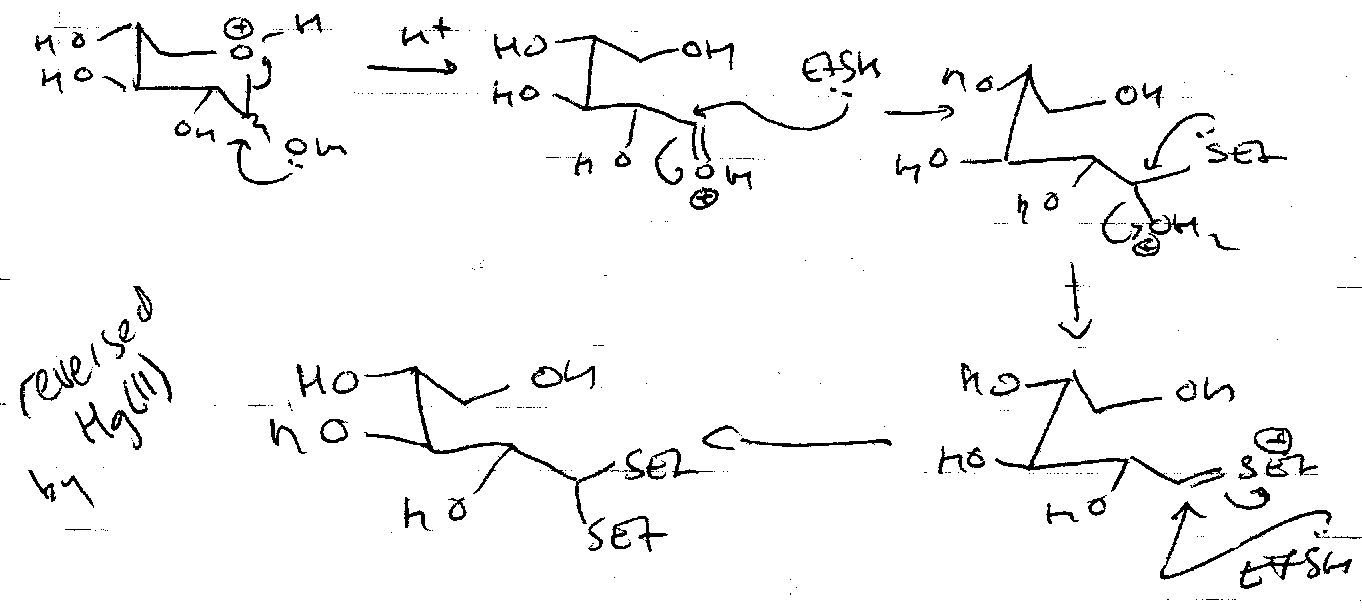
Important Acetal Mechanisms
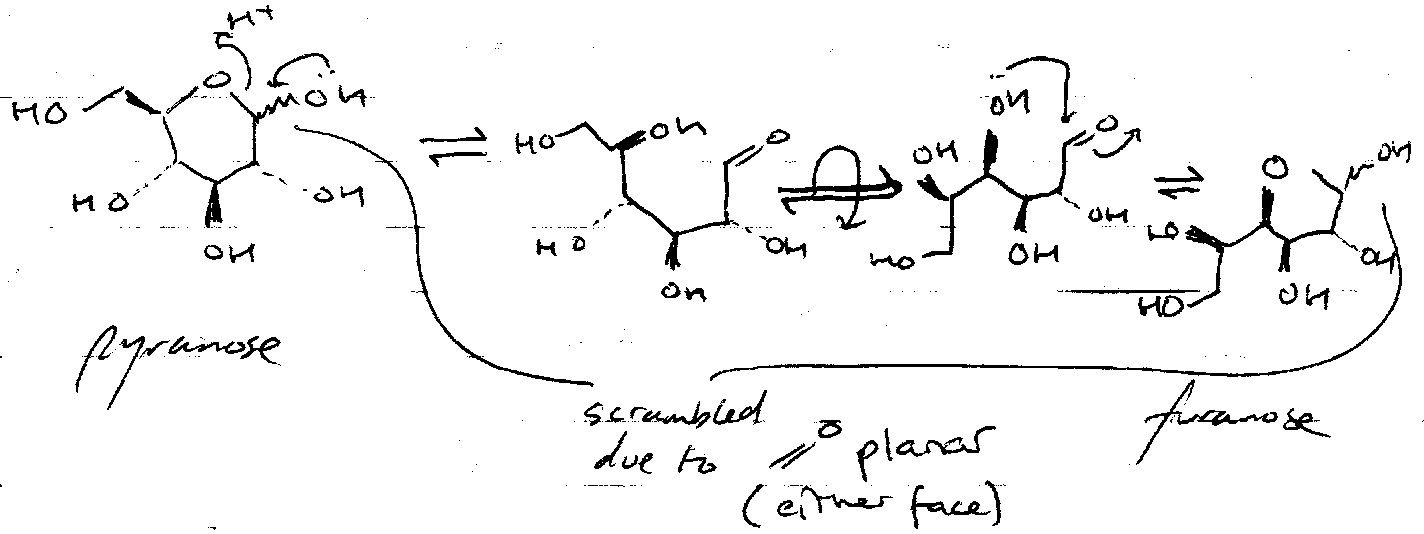
Hemiacetals exist in equilibrium with aldehyde and alcohol. For monosaccharide rings, this lies almost entirely on the side of the hemiacetals. The interconversion is catalysed by acid and base.
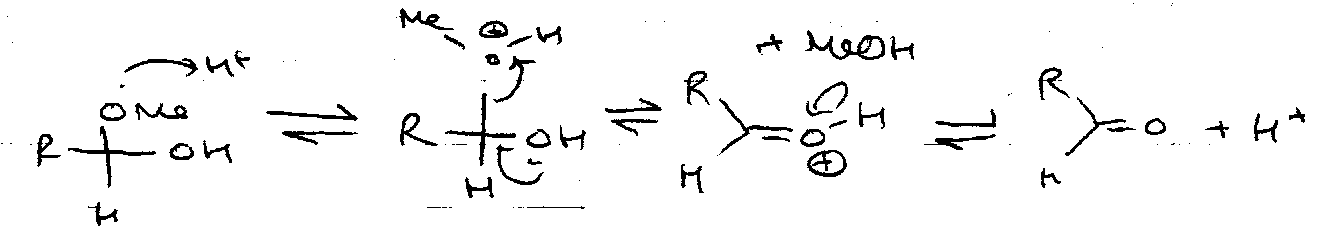
Acid:
Formation –
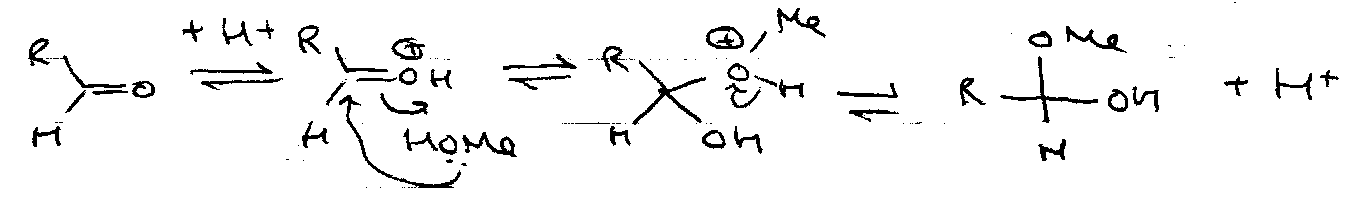
Hydrolysis –
Base:
Formation –
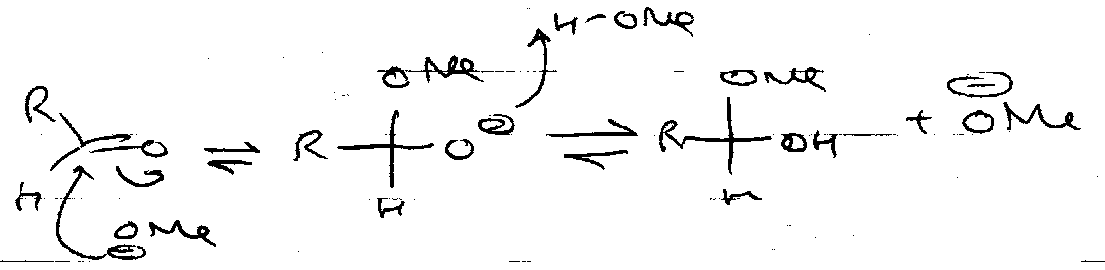
Hydrolysis –
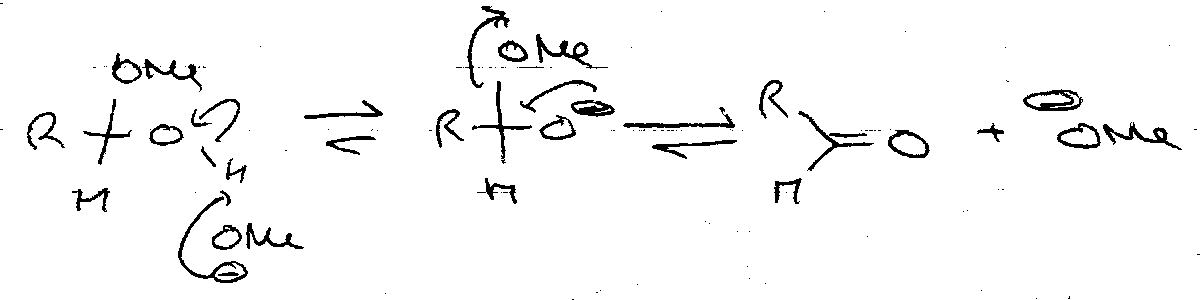
In solution, the following equilibrium exists:

Acetal Formation
Acid catalysed only now. Need to turn OH into a leaving group.
Fischer Glycosidation
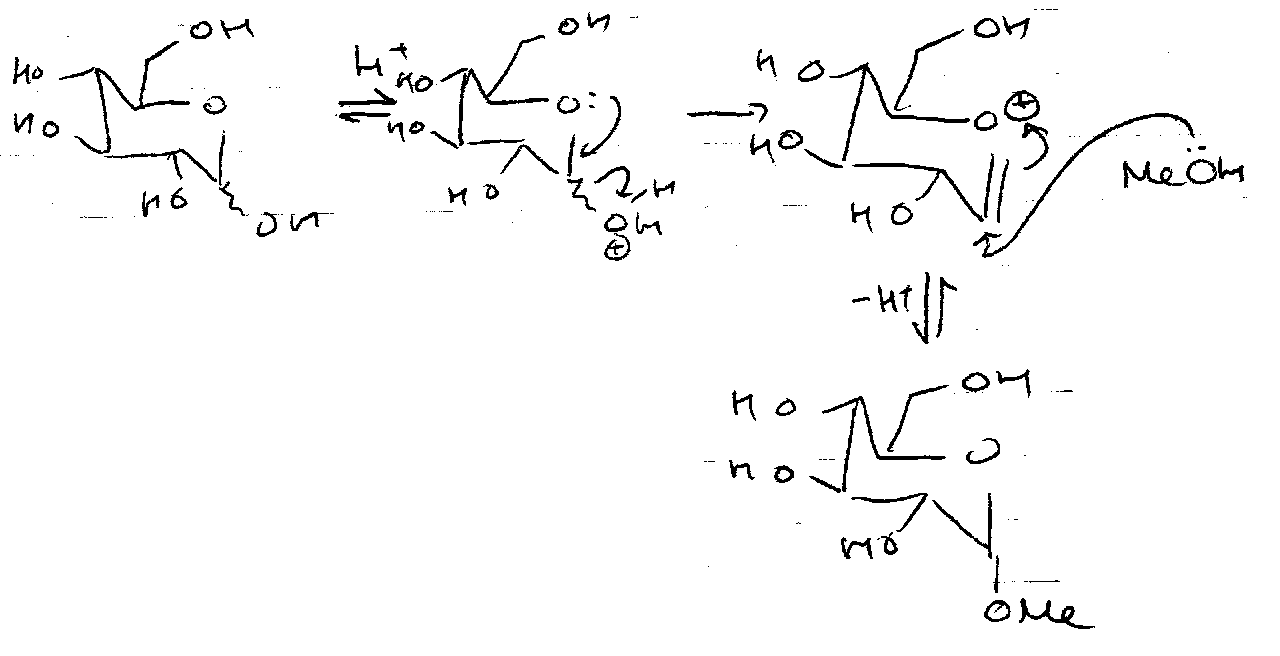
Thermodynamic Control → axial OMe, due the anomeric effect. Only reversed by H+ treatment → effectively protected anomeric centre as a glycoside.
The Anomeric Effect
Electronegative substituents prefer to be axial. Why?
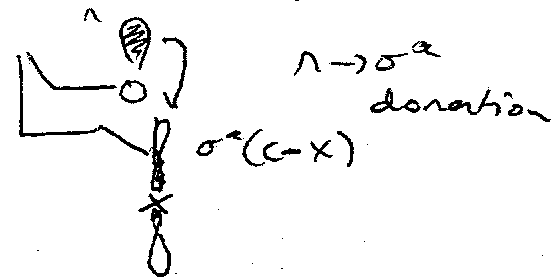
Antiperiplanar alignment required for the above stabilising donation.
Mutarotation
Change in optical rotation between α and β positions. Equilibrium process.
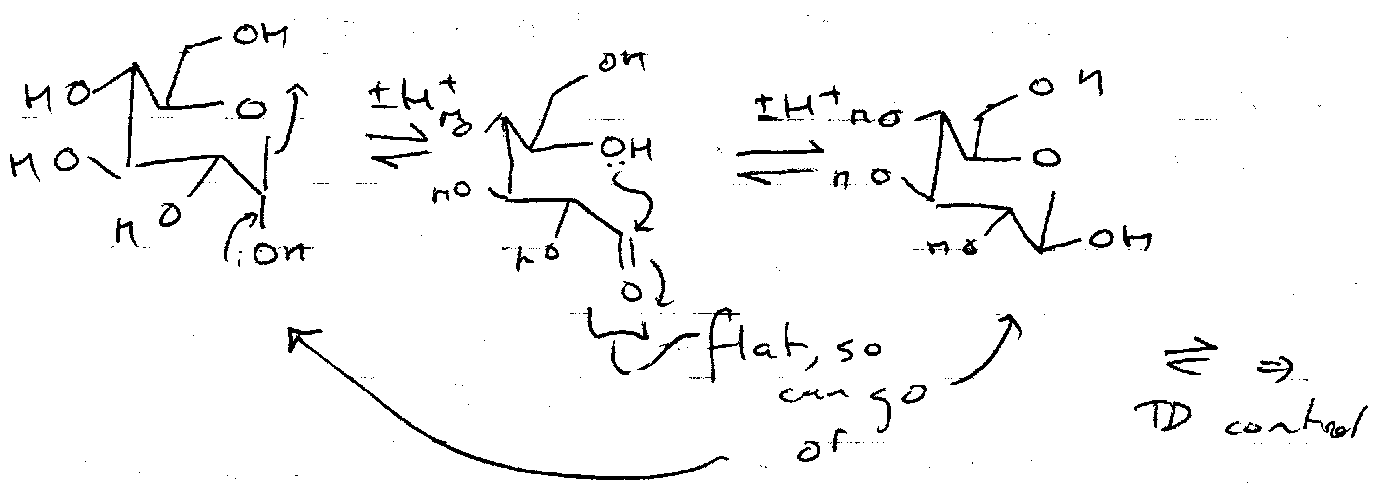
This is a competition between the (electronic) anomeric effect (axial) and sterics (equatorial). The β (equatorial) form usually dominates due to solvation.
Reactions
3 regions of reactivity in a sugar molecule:
1) Primary CH2OH (x1)
2) Secondary CHOH (x3)
3) Anomeric OH (x1) – complex reactions (so often protect it). Optical rotation, OH nucleophilic and carbonyl-like properties.
Acetylation
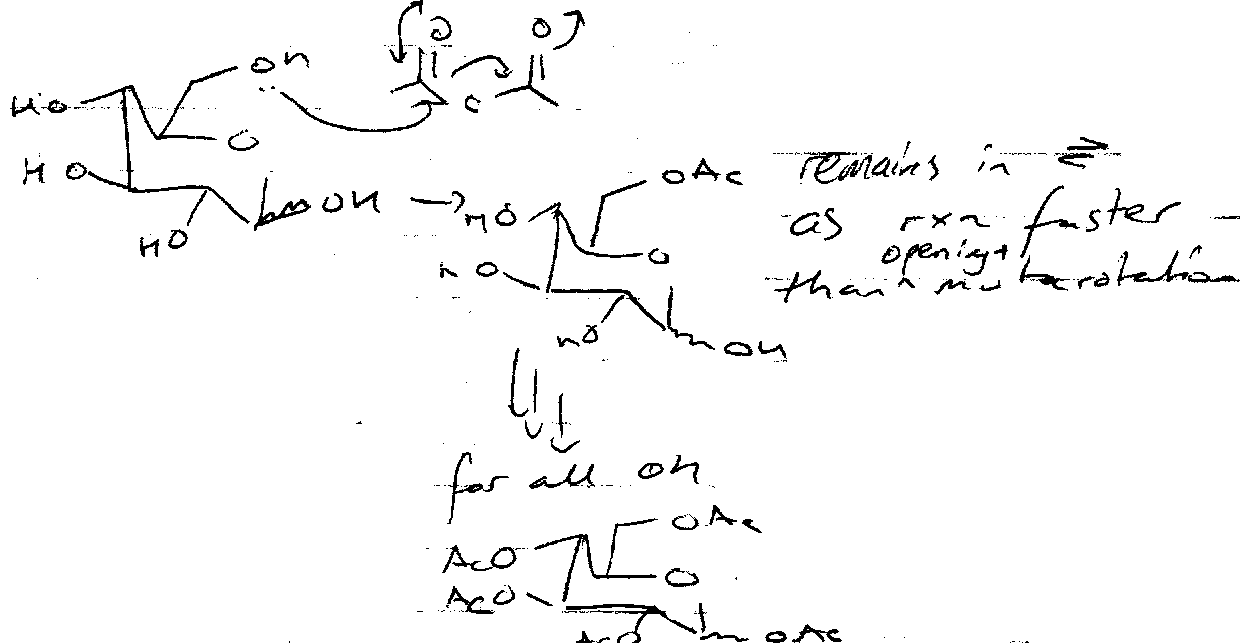
Reaction slower with Ac2O + NaOAc, and usually selects β product as mutarotation has time to occur. Equatorial OH (β) much more nucleophilic than axial (α), therefore it dominates.
Reaction with Lewis Acid + Ac2O favours α. Lewis Acid catalyses equilibrium of α and β and α-anomer is thermodynamically favoured.
Protecting Groups
Require:
- Good yield.
- Stable to other reaction conditions.
- Readily removed under appropriate conditions.
Allows for selective access to the desired OH.
Acetals – see later. Anomeric Centre protection.
Ether Protection
Benzyl – alcohol + Benzyl Halides + base (NaH). Simple SN2 displacement of Hal. Catalysed by Iodide (nucleophilic catalyst).
Removed by mild catalytic hydrogenation, e.g. Pd, C, Ni.
Also paramethoxybenzyl (PMB). Selective, and removed using DDQ.
Trityl Ethers
Very selective for primary OH. Bulky. Ph3CCl + pyridine to put it on. Cleave with mild H+ (e.g. after protecting secondary OHs with BnBr).
Silyl Ethers
Silyl chloride + weak base (imidazole). TMSCl protects all OH. TBDPSCl & TBDMSCl regioselectively protect the primary OH.
Removal is achieved with acid or more likely F- (Bu4NF) as this will not deprotect other groups that are acid labile.
Ester Protection
Benzoyl
Benzoyl Chloride + amine base. Removed using a nucleophile such as methoxide (transesterification).
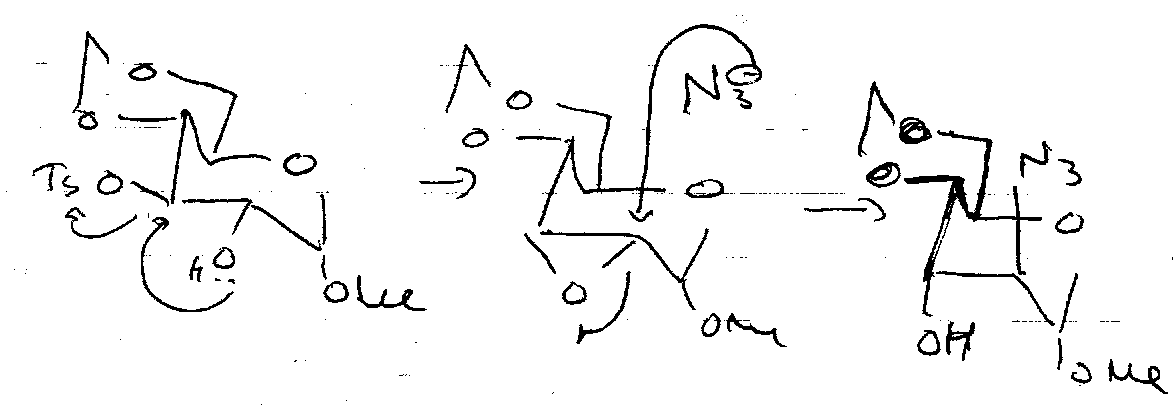
Sulphonate Esters
Protect and good leaving group. Put on using TsCl + pyridine.
Nucleophilic Substitution
Need to convert OH into a leaving group. The mechanism is usually SN2, so expect inversion. SN2 is likely due to the electron withdrawing groups β to OH (present for all but the primary alcohol) destabilises C+ in SN1.
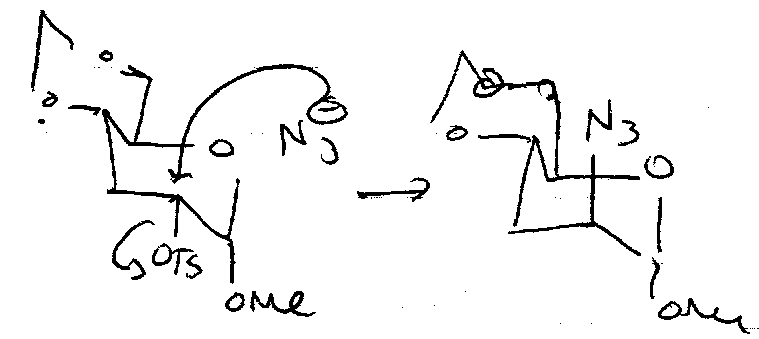
Also, epoxide formation:
Oxidation & Reduction
Oxidation
Usual reagents are PCC, Swern, Moffatt. Cleavage – use IO4-.
Reduction
Often make xanthate (CS2, NaOH, MeI), then Bu3SnH + AIBN.
Reactions at the Anomeric Centre
Interchange between furanose and pyranose forms is important here:
Nucleophilic Substitution
SN1 –
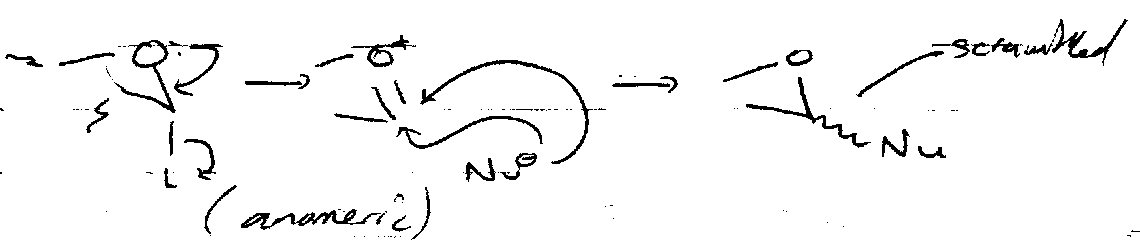
SN2 –
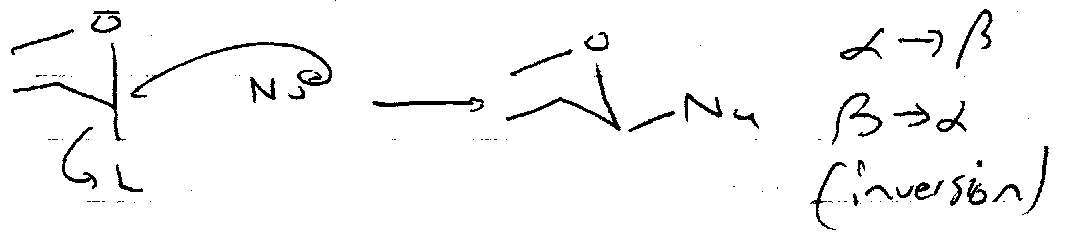
Stereocontrol often provided by Neighbouring Group Participation.
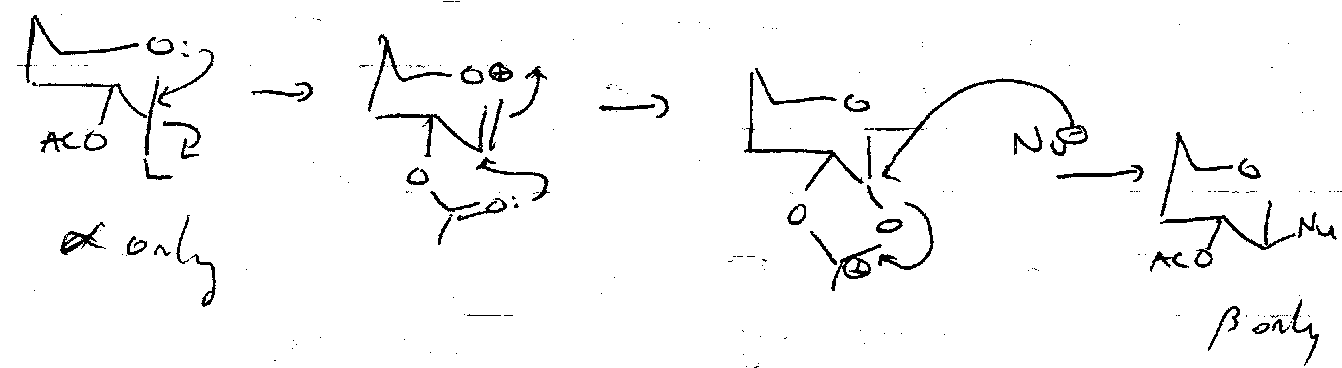
Acetates
AcOH + H+ (or Lewis Acid + Nucleophile). Good Leaving Group.
α-anomer only due to equilibrium, such that thermodynamic stability overrides neighbouring group participation. If F-/Cl- introduced, C-X bond strength increase and equilibrium is slowed, so see some β-anomer.
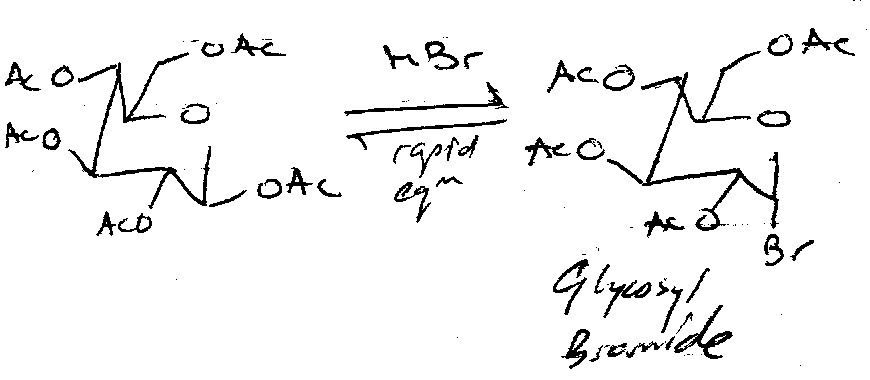
Glycosyl Bromides
Undergo many reactions.
Nucleophilic Substitution can be carried out to make thioglycosides or azidoglycosides. Ag(I) and Hg(II) (halophiles) are often used to speed up the removal of Br, especially with poorer nucleophiles, e.g. in disaccharide synthesis (ROH – the other sugar – is a weak nucleophile).
Also,
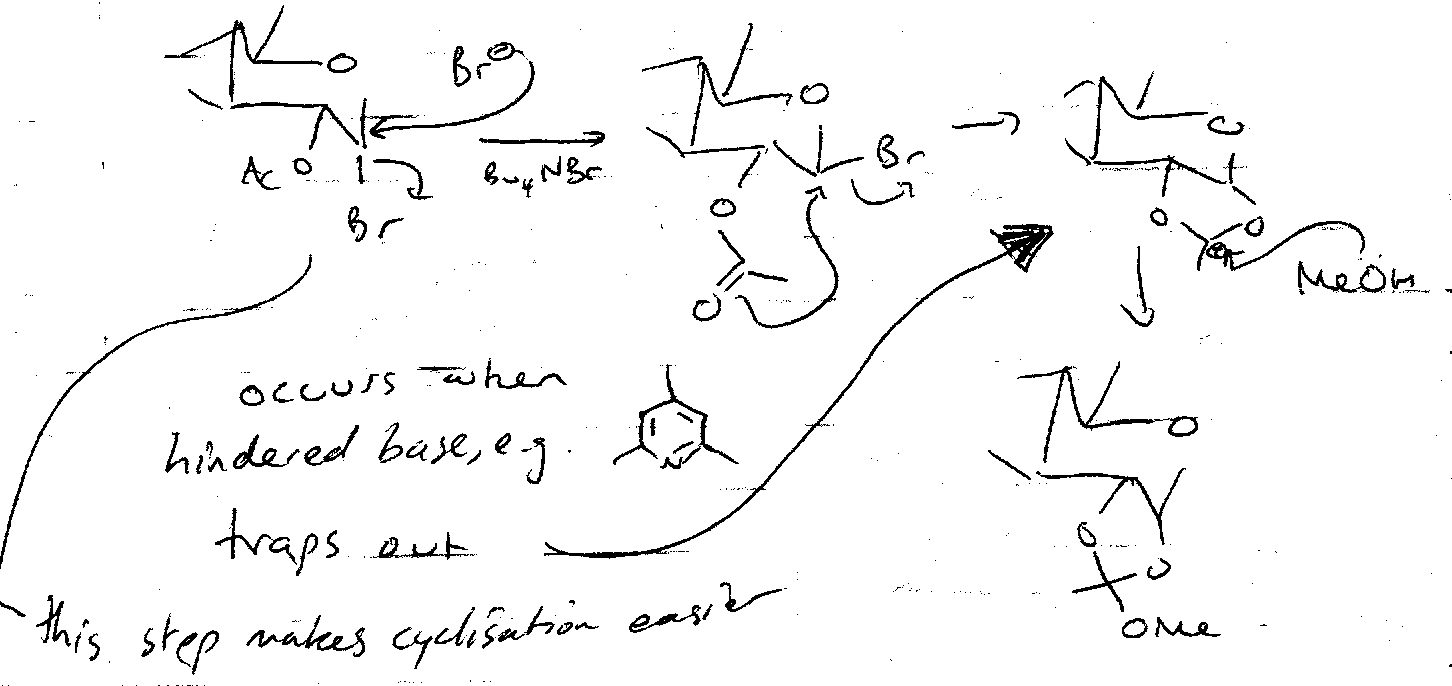
Reductive Elimination
Makes the Glycal.
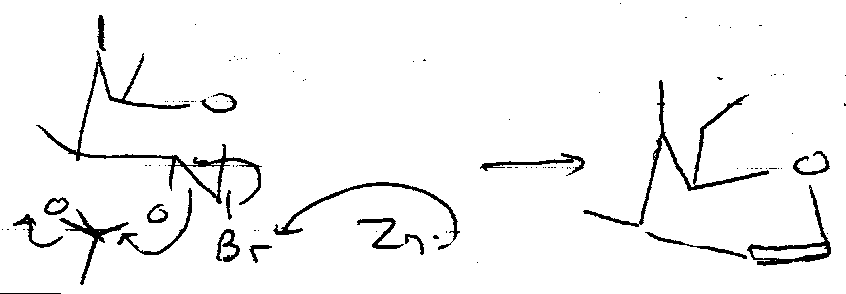
Free Radical Reactions
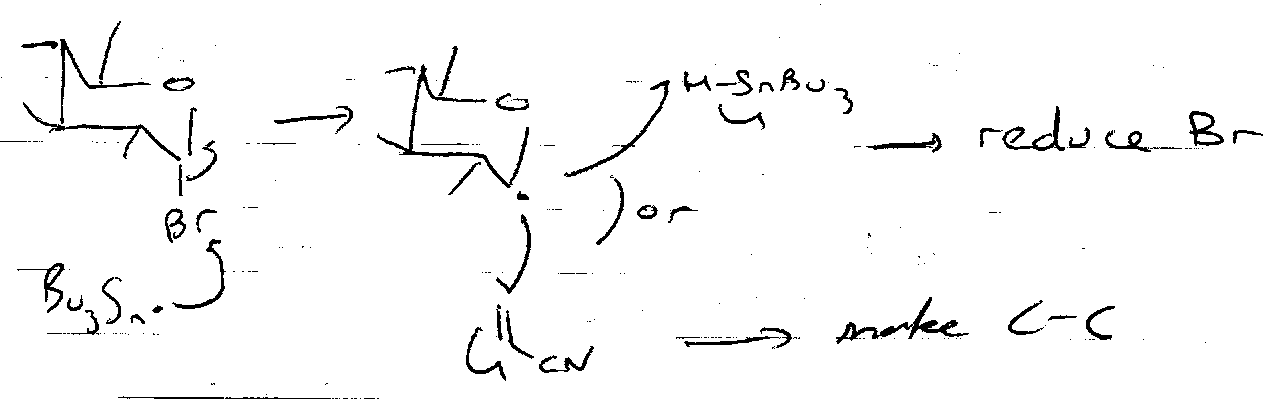
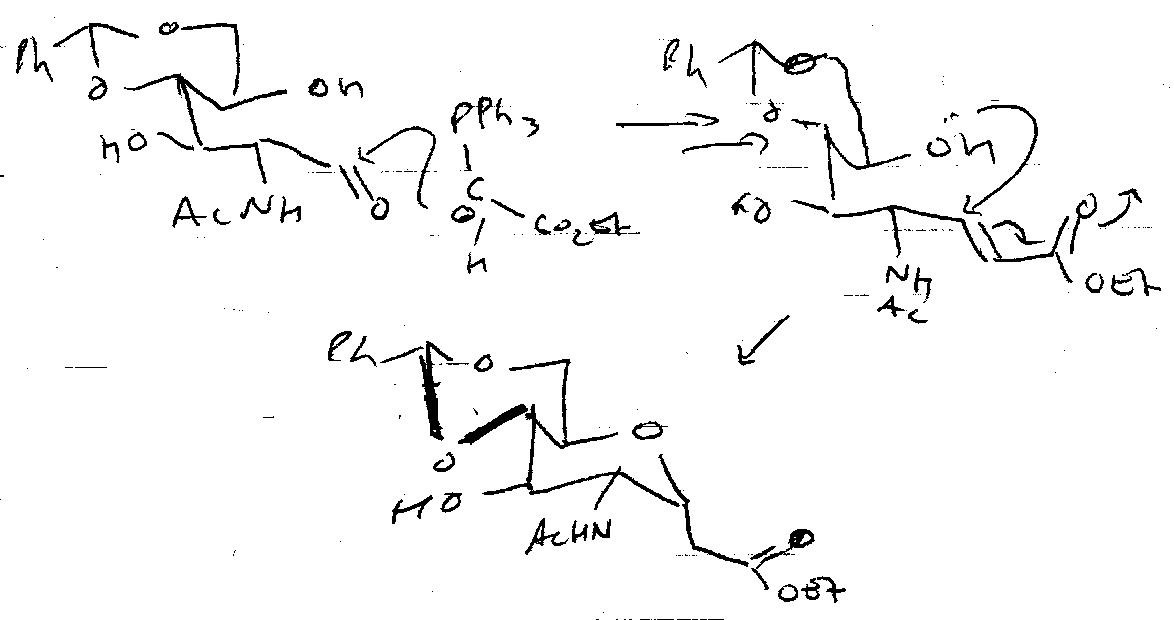
Nucleophilic Addition
This is used to capture the open chain aldehyde. Useful for forming C-C at anomeric centre (lengthening).
Dithioacetal –
Wittig (& modifications) –
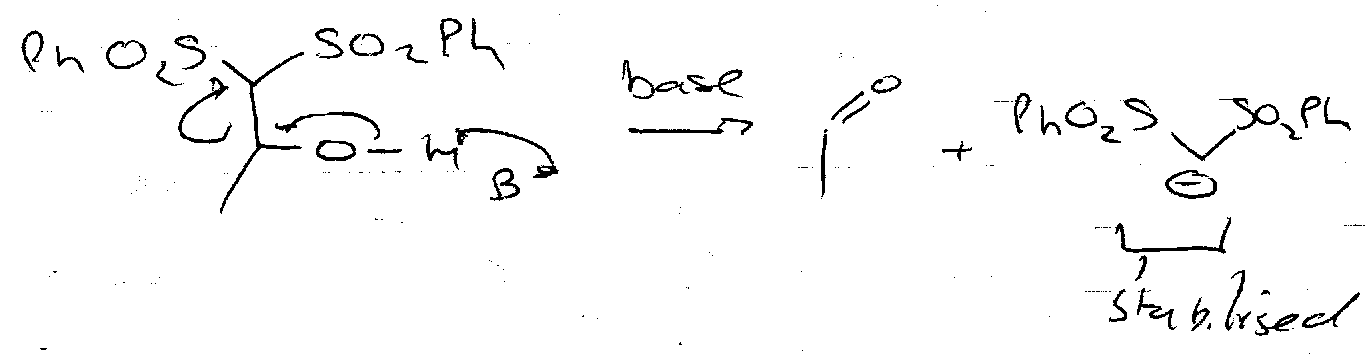
Chain Degradation
Use dithioacetal (above), and react with MCPBA (SPh → SO2Ph). Forms a leaving group on terminal carbon (originally the anomeric centre).
Chain Extension
Kiliani Ascension: treat free sugar (aldehyde form) with cyanide.
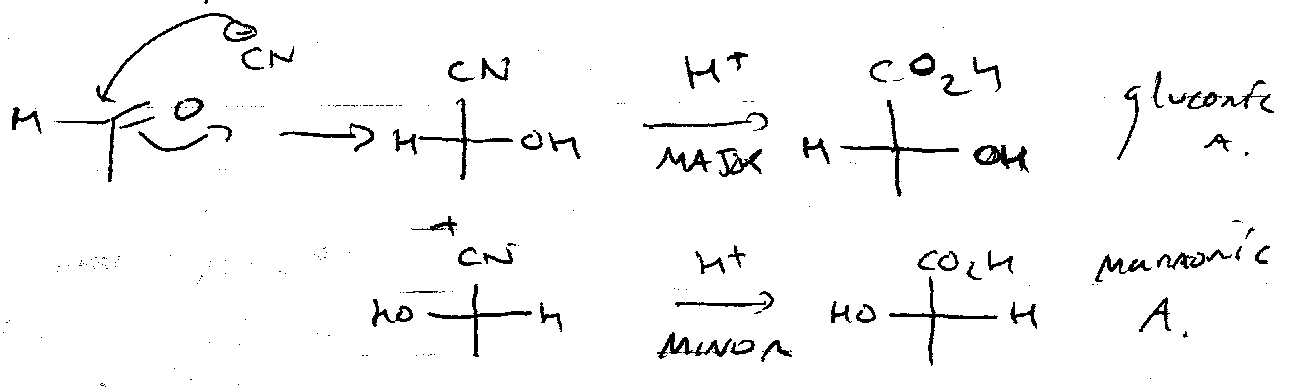
There are similar methods with CH2NO2 (Nef Reaction) and CH2N2 (diazomethane).
Oxidation

Reduction

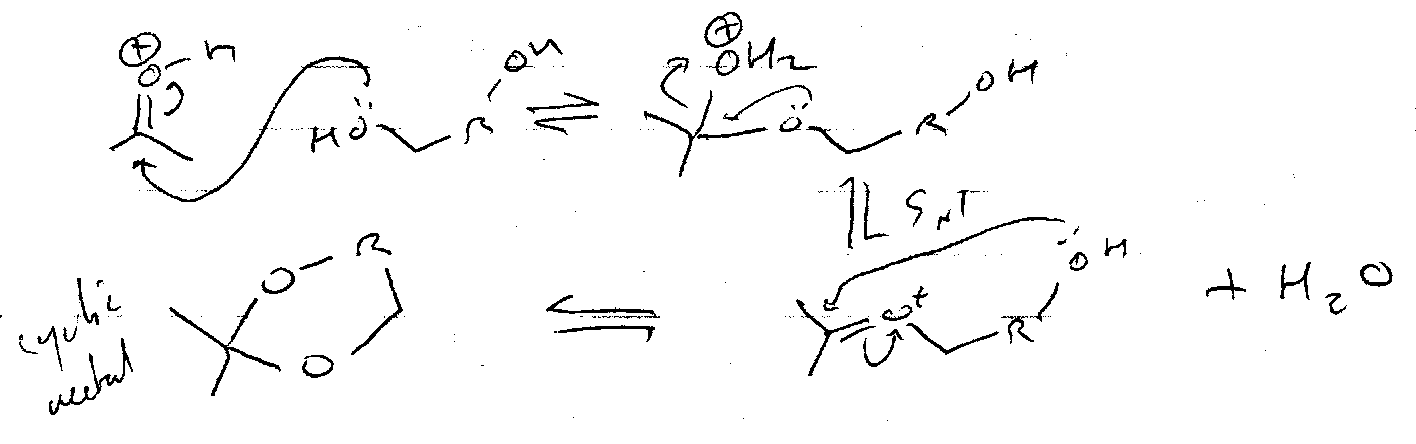
Cyclic Acetals
Commonly formed to selectively protect some of the secondary hydroxyl groups.
Mechanism:
Ring Sizes –
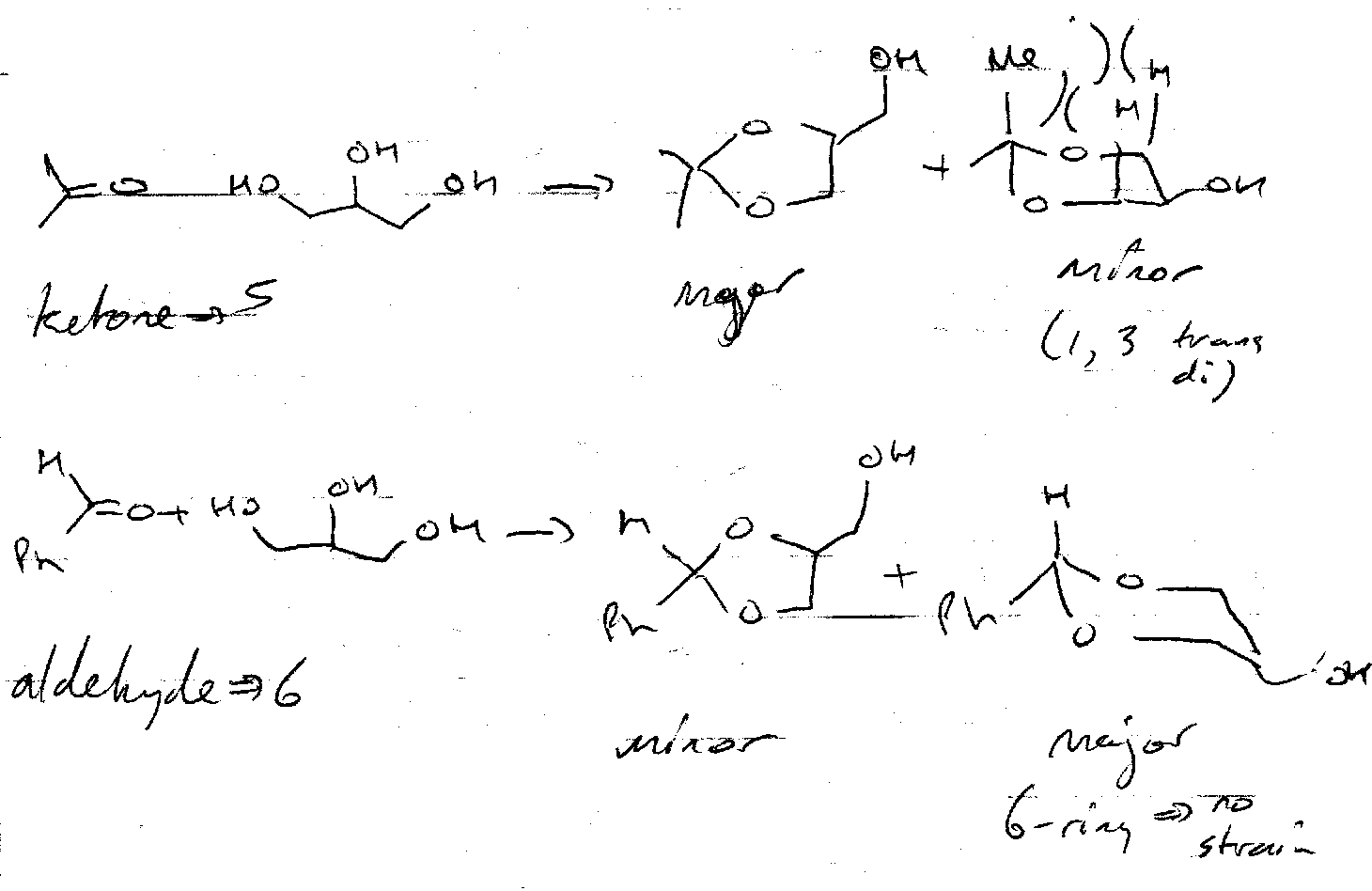
This is only a guide, however.
Acetonide Protection –
Acetone forms five-membered rings to protect 2 x OH. These must be cis (trans-fused ring too strained), and so is selective. It is acid catalysed.
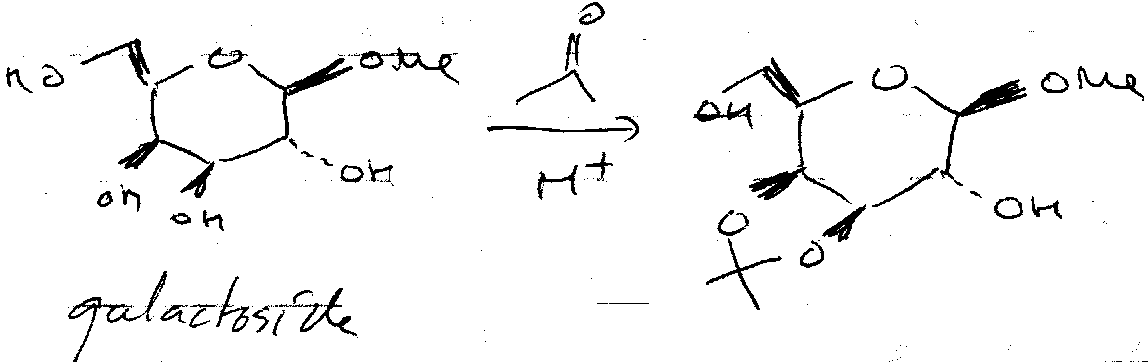
Reaction with glucose is slightly different. Glucose can only form one acetonide ring using the anomeric centre + C2. But, H+ means that equilibrium with the furanose form exists. This can react with two acetone molecules (one end is not part of the ring, so cis requirement is lost).
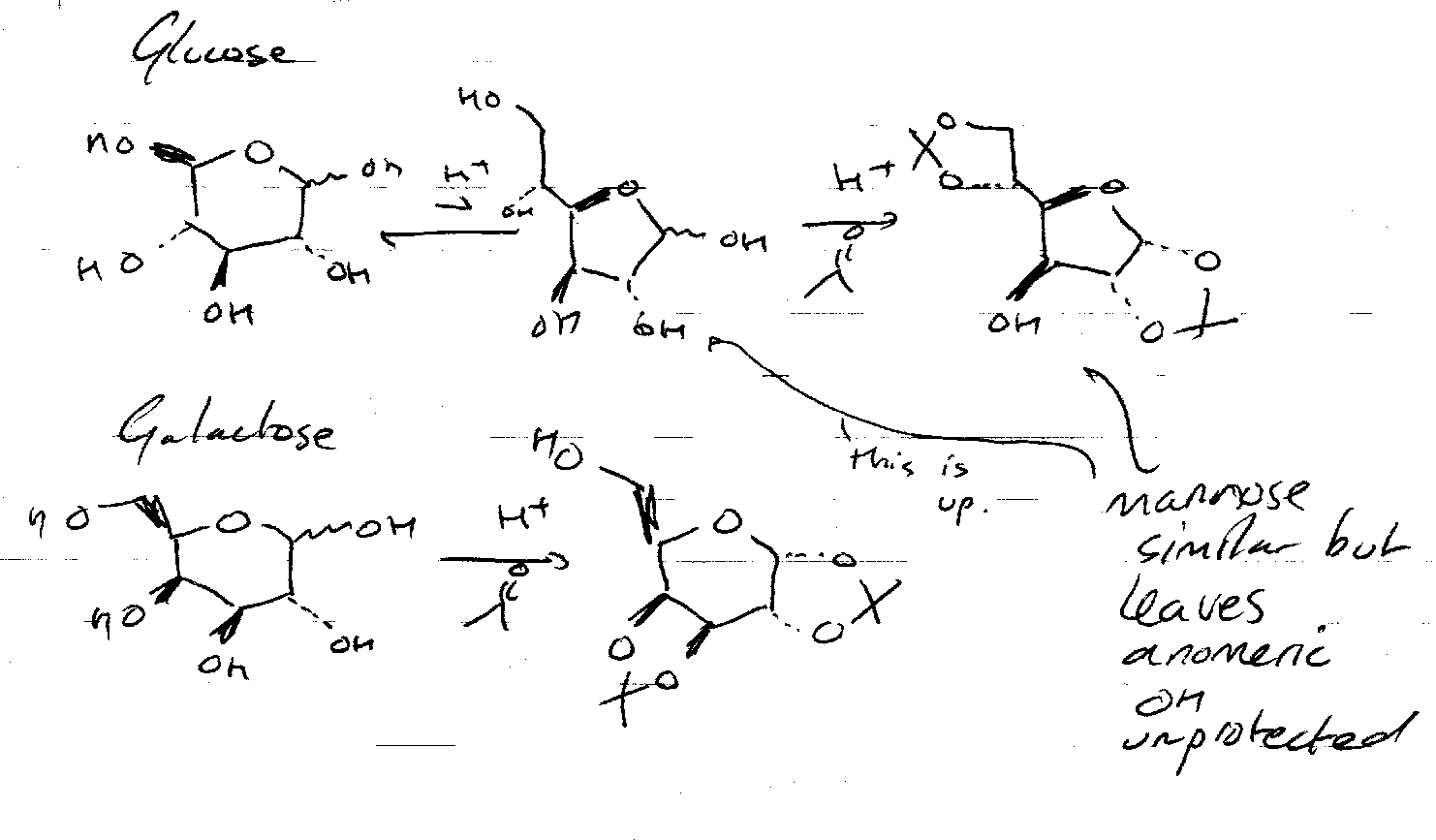
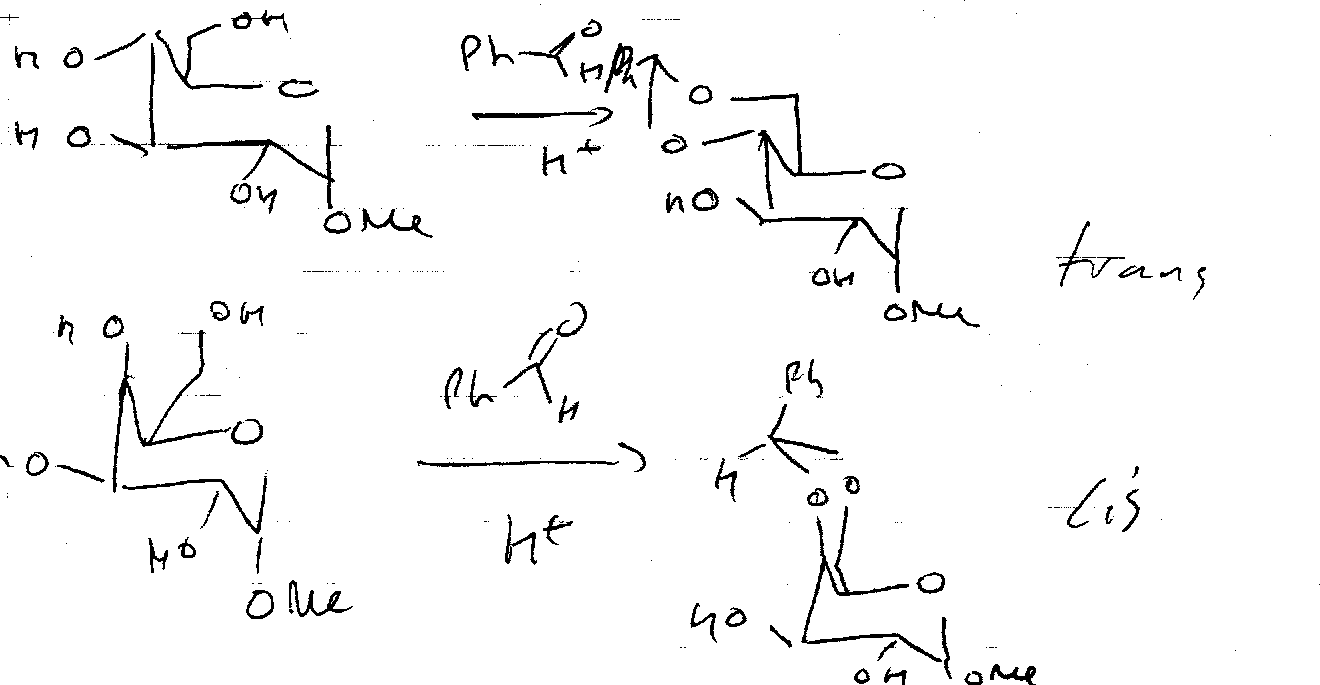
Benzylidene Protection –
Benzaldehyde is used instead of acetone. This forms 6 membered rings.
Deprotection can be achieved in both Acetonide and Benzylidene Protection by forcing the equilibrium back, i.e. reversing acetal formation. Hence, AcOH / H2O is used (pushes back to carbonyl by hydrolysis). Selective removal of just the primary acetonide is achieved by less harsh conditions.
I am happy for them to be reproduced, but please include credit to this website if you're putting them somewhere public please!