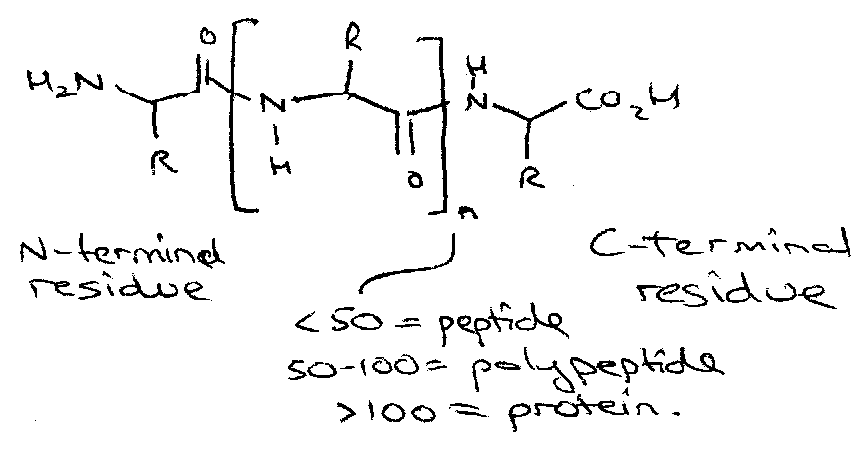
Peptides
Advanced Level Only - not too many mechanisms either as they're easy but I've covered most of the protection and conversion options and structure determination. Lots from the Primer, but filled in the bits it missed.
Peptides Notes
The Basics
The Proteinogenic α-amino acids
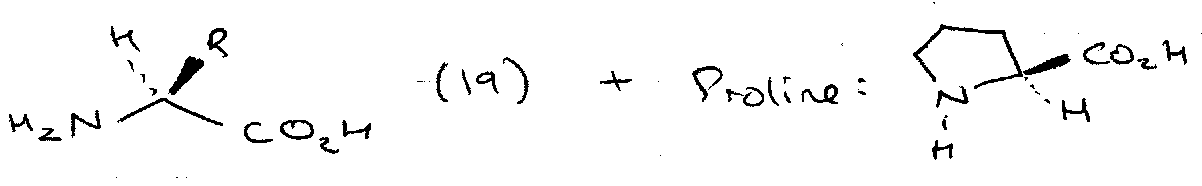
|
Amino Acid |
Abbreviation |
Side Chain (R) |
|
Glycine |
Gly |
H |
|
Alanine |
Ala |
Me |
|
Valine |
Val |
CHMe2 |
|
Leucine |
Leu |
CH2CHMe2 |
|
Isoleucine |
Ile |
CH(Me)CH2Me |
|
Phenylalanine |
Phe |
CH2Ph |
|
Tyrosine |
Tyr |
CH2(4-hydroxyphenyl) |
|
Tryptophan |
Trp |
CH2(3-indolyl) |
|
Methionine |
Met |
CH2CH2SMe |
|
Cysteine |
Cys |
CH2SH |
|
Serine |
Ser |
CH2OH |
|
Threonine |
Thr |
CH(Me)OH |
|
Arginine |
Arg |
CH2CH2CH2NHC(=NH)NH2 |
|
Lysine |
Lys |
(CH2)4NH2 |
|
Histidine |
His |
CH2(4-imidazolyl) |
|
Glutamic Acid |
Glu |
CH2CH2CO2H |
|
Aspartic Acid |
Asp |
CH2CO2H |
|
Glutamine |
Gln |
CH2CH2CONH2 |
|
Asparagine |
Asn |
CH2CONH2 |
Peptides
What they do:
Hormones – insulin and oxytocin.
Antibiotics – penicillin.
Drugs – zoladex (prostate cancer), crixivan (AIDS).
Structure Determination
Usually by chemical degradation, although Physical Methods can also be used.
- Oxidise S-S bridges with peracid.
- C-terminal: hydrazinolysis
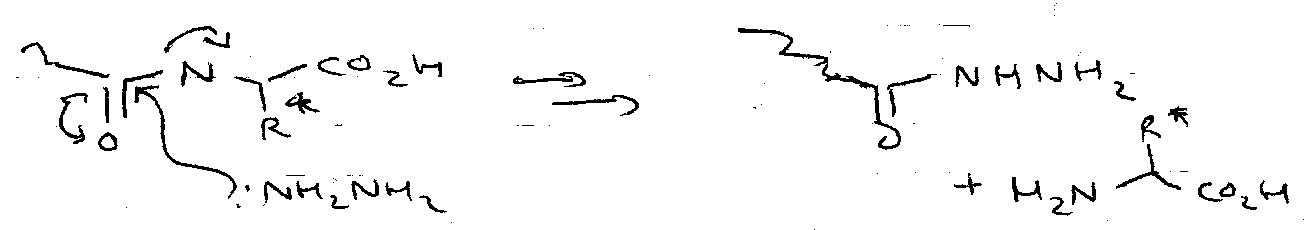
- N-terminal:
Sanger Method –
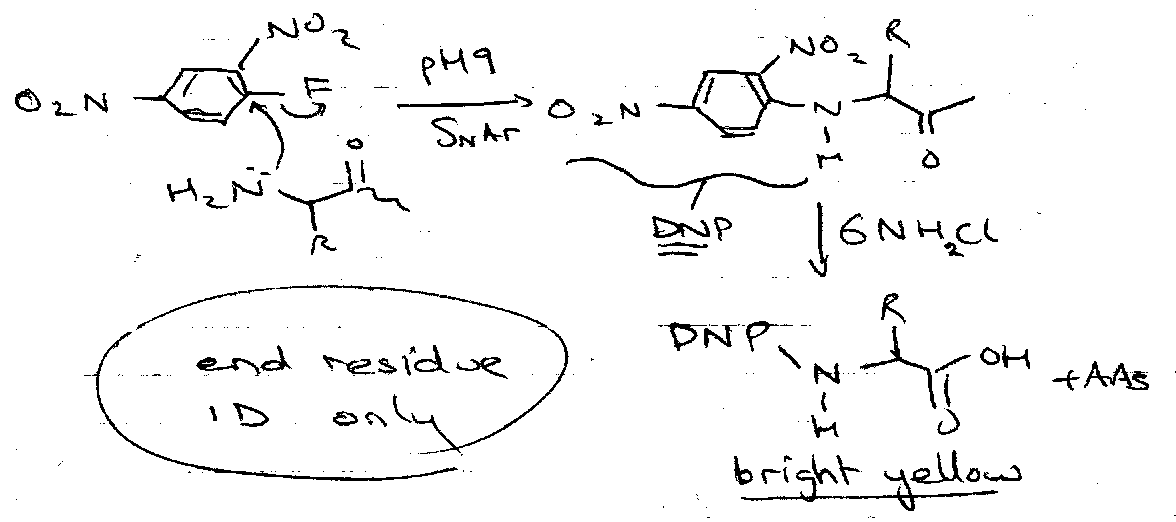
Edman Method –

- Selective Fragmentation – add BrCN at Met residues.
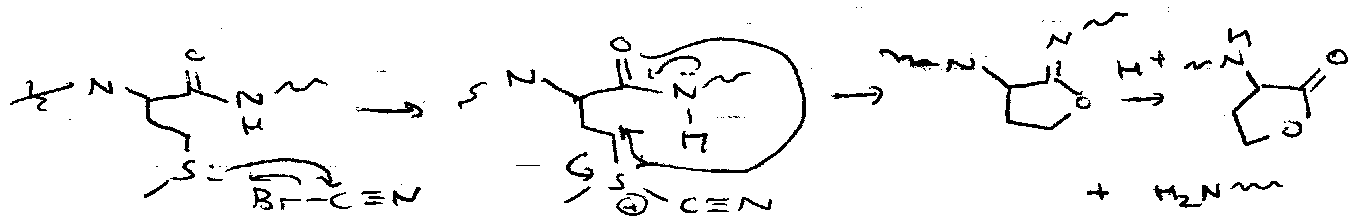
- Protease Enzymes, e.g. trypsin (cleaves basic side chains – Lys, Arg).
4 & 5 are often combined – overlap sequences and thus helps structure assignment.
Synthesis Overview
Unsymmetrical dipeptides:
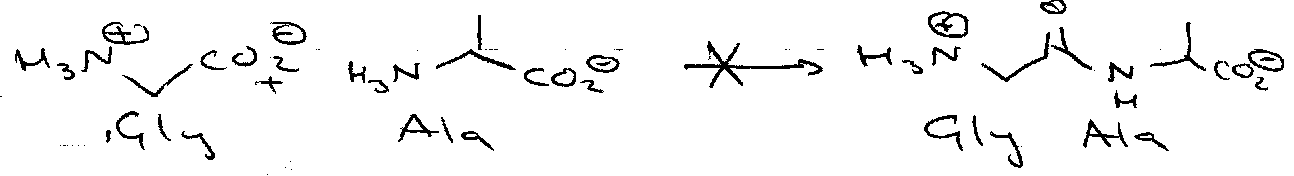
Need to protect them selectively –
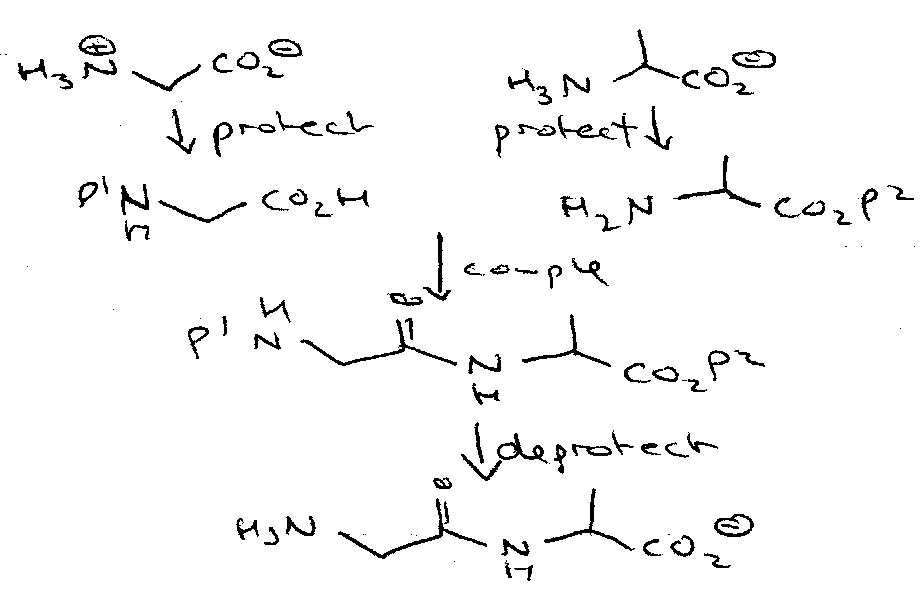
Depicted as:
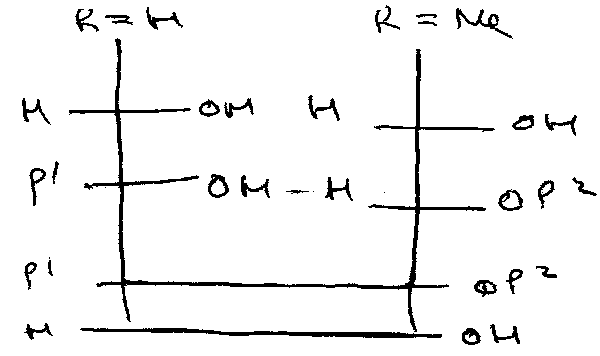
Protecting Groups
- Easy to introduce.
- Protect Functional Group during amide formation.
- Removable without harming rest of the molecule.
- No loss of stereochemistry in 1-3.
α-amino Protection
Alkoxycarbonyl Protection
Also known as urethanes or carbamates. They are both amides and esters. As amides, they have low nucleophilic reactivity at nitrogen. As esters, they have relatively low reactivity wrt acyl-oxygen fission, but they can be degraded alkyl-oxygen fission to carbamic acids, which decarboxylate spontaneously under practically all conditions, regenerating the parent amine.
They are also good because they are nearly immune to racemisation.
Benzyloxycarbonyl (Z)
Less reactive acylating agents (i.e. not the chloroformate) are sometimes preferred to prevent dipeptide formation:
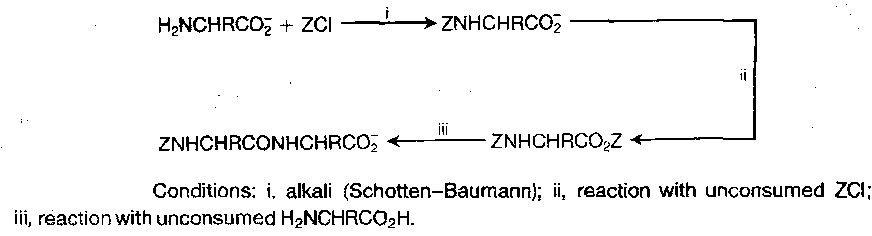
Hence use reagents such as:
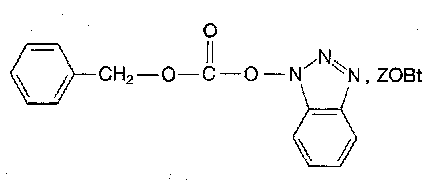
Mildly basic or nucleophilic reagents do not generally affect the Z group at ambient temperature. The main exception to this is hydantoin formation:
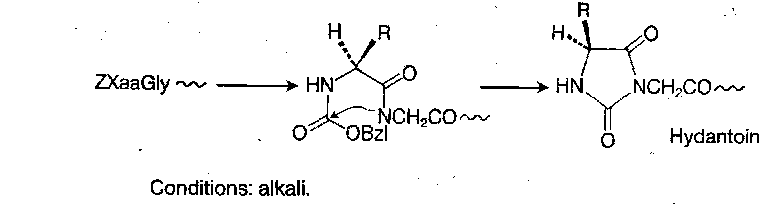
However, this is only really significant with ZXaaGly-peptides.
Mildly acidic conditions (e.g. half an hour with neat TFA in an ice-bath) are also without effect. This allows it to be carried through synthesis with more acid-labile protecting groups (orthogonality).
Cleavage conditions are HBr/AcOH or catalytic hydrogenolysis. HBr/AcOH proceeds by an SN2-like mechanism:

The conditions are quite severe though, but most peptides survive it undamaged, unless susceptible side-chains are present. This is usually due to the generation of electrophilic species such as benzyl cations which attack electron-rich side chains (e.g. Trp, Tyr, Met). Additives like anisole and DMS diminish this difficulty by acting as “scavengers” for such species. Increasing the acidity of the conditions tends to favour an SN1 mechanism, where electrophile generation is more of a problem. Typical Scavengers are triethyl silane (Et3SiH), which is attacked by CF3COO- and donates its H to the electrophile, converting it to a harmless hydrocarbon.
Catalytic hydrogenolysis is a useful alternative providing S side-chains are not present, as the sulphur contaminates the catalyst.
t-Butoxycarbonyl (Boc)
t-butyl chloroformate decomposes too easily to prepare Boc derivatives. The azide was used for some time, but now the anhydride is preferred (less explosive). The only disadvantage with this is cost.


The Boc group is completely stable to catalytic hydrogenolysis conditions, but is much more acid labile than the Z group, so it is highly orthogonal to this.
Basic and nucleophilic reagents have no effect at all on the Boc group, even on prolonged exposure. No hydantoin formation.
Removal is facile with TFA (either neat or diluted) at ambient temperature for about half an hour.

The t-butyl moiety ends up as isobutene unless it is trapped by a nucleophile, so scavengers are often added (these are essential if sensitive residues such as Trp are present). Mineral acids attack Boc groups rapidly, and must be avoided in acidification and wash procedures (use citric acid wash – weak acid and does not extract into organic solvents from water).
2-(4-biphenylyl)-isopropoxycarbonyl (Bpoc)
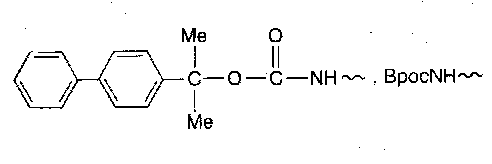
Even more acid labile than Boc (biphenylyl adds stabilisation to carbocation generated). Can remove in the presence of Boc by brief treatment with chloroacetic acid-dichloromethane mixtures. It is also cleaved by catalytic hydrogenolysis.
9-fluorenylmethoxycarbonyl (Fmoc)
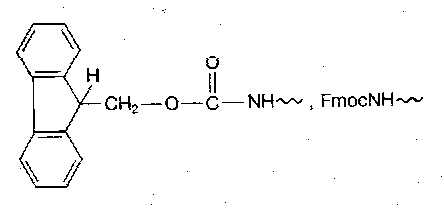
Introduced in a Schotten-Baumann manner, using FmocCl. Dipeptide formation is a risk, so several less reactive reagents such as FmocOSu may be used.
Fmoc is very stable to acid, but cleaved swiftly under certain basic conditions. Piperidine (20% in DMF) is the routine reagent, but fluoride ions in DMF also work. The mechanism is very rapid, and proceeds via E1cb via the stabilised anion:
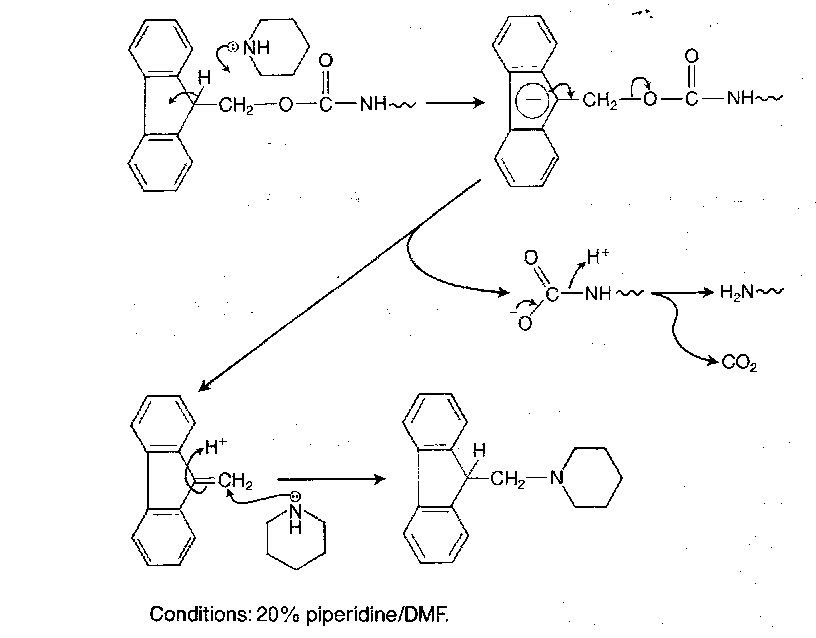
This does not affect Boc or Z groups, or most other protecting groups in fact. Fmoc is not completely inert to catalytic hydrogenolysis, so this should be avoided for Z removal in the presence of Fmoc.
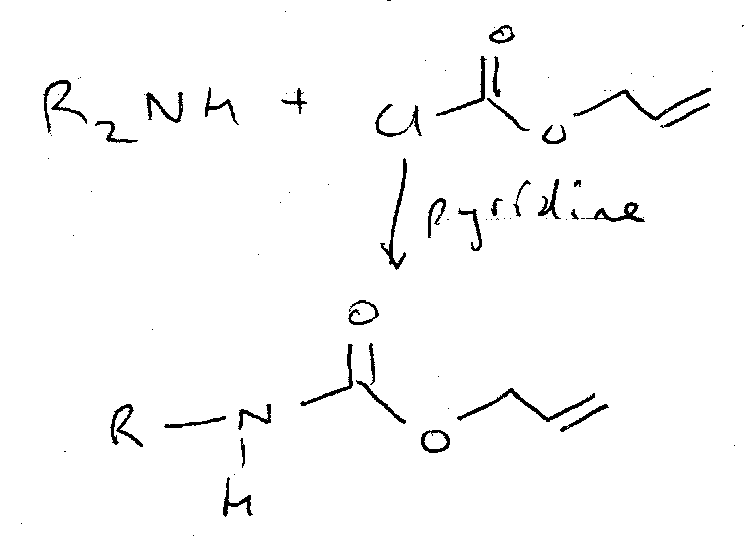
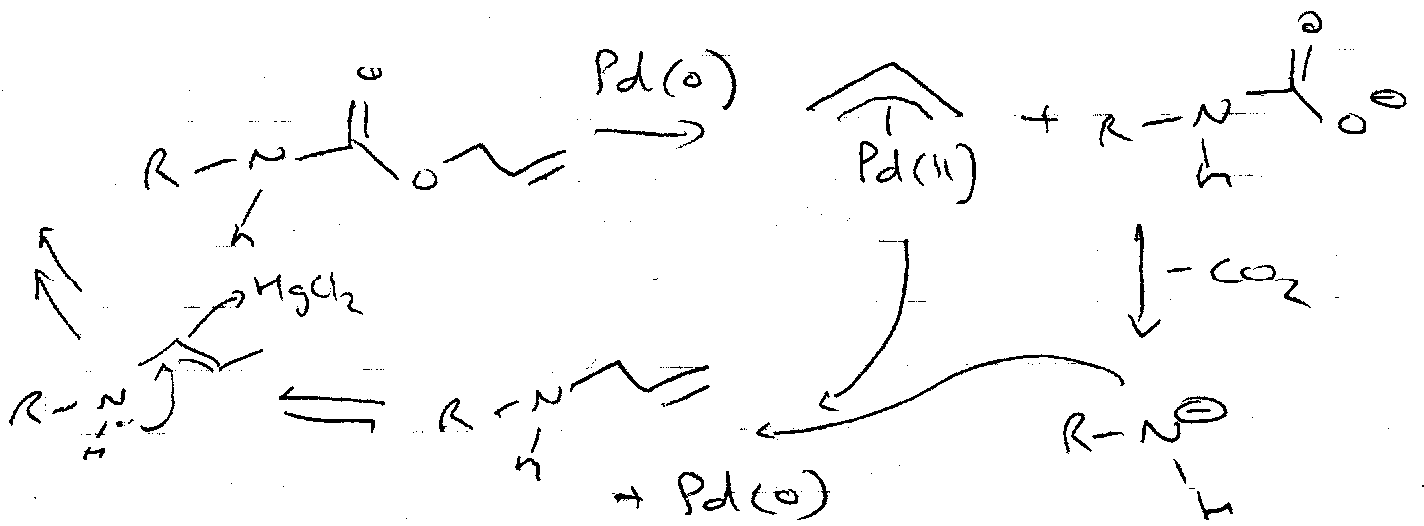
Alloc
This is another orthogonal option. It is added using the chloroformate, but usefully can be removed by Pd catalysts as shown.
Other Methods
There are several useful protecting groups that are not alkoxycarbonyl based.
Triphenylmethyl (trityl, Trt)
Reaction with α-amino esters gives better yields than Trt-Cl directly. However, this is sluggish, so the following method is usually used:
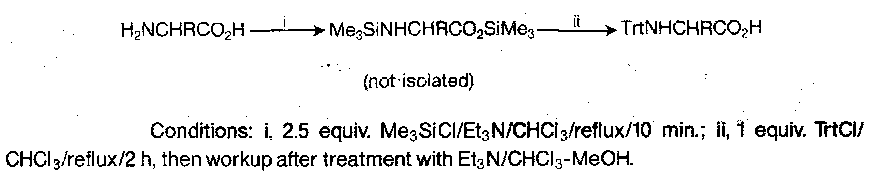
The trityl group is very stable to base, very labile to acid (acetic acid of pH 4 works), and does not survive catalytic hydrogenolysis.
Only problem is the extreme bulk can cause difficulties in peptide bond formation.
2-nitrophenylsulphenyl (Nps)
NpsCl + amino acid are not very stable in the free acid state, so they are generally isolated, purified and stored as dicyclohexylammonium salts. They can be liberated by cautious acidification with sulphuric acid. Nps group is stable to mild base, but not to acid or catalytic hydrogenolysis.
Sulphenamides deactivate the nucleophilic reactivity at the amino nitrogen by S-N π-bonding effects, and it is backed up the electron withdrawing NO2 substituent.
Deprotection proceeds as follows:
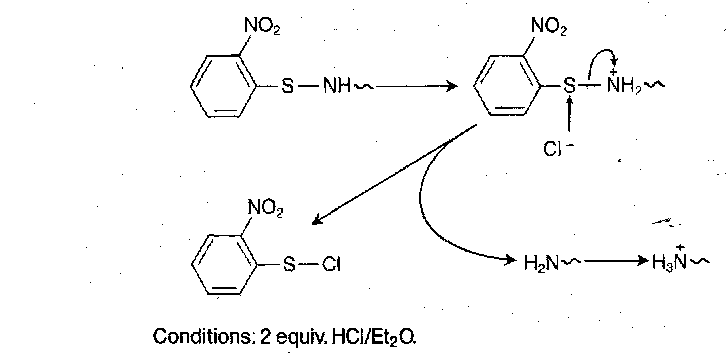
Dithiasuccinoyl (Dts)
Add EtOCS2Me, then ClSOCl, then aqueous acid to amino ester. The chemistry is complicated (and smelly) but it has one advantage – it can be selectively removed by treatment with thiol RSH and a mild base such as Et3N.
α-Carboxy Protection
The carboxy does not always need protecting (it is less nucleophilic than the amine) but often is for solubility advantages and product isolation. The usual means of protection is esterification, and the carboxy group can be regenerated on alkyl-oxygen fission.
Esters
Methyl and Ethyl Esters
Amino acids react with hot methanolic hydrogen chloride. Can also use thionyl chloride + methanol. They provide good protection, surviving most peptide bond forming procedures, but as a consequence are hard to remove. For example, HBr/AcOH does not affect them, nor does catalytic hydrogenation. Saponification is sometimes satisfactory but has the risk of racemisation and side reactions such as hydantoin formation. Hydrazinolysis can also be used, but not entirely reliable either.
Side reactions are also possible at the dipeptide stage, because dipeptide methyl ester free bases cyclise to diketopiperazines (DKPs) rather easily:
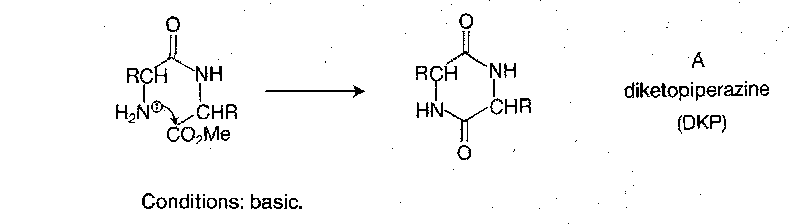
Benzyl Esters
Amino Acid + TsOH + BzlOH over heat gives H3N+CHRCO2Bzl. Driven by removal of water. Isolated as salts, as the free bases are unstable (and prone to DKP formation, above).
They can be cleaved by hydrazinolysis or saponification, but also by catalytic hydrogenolysis, HBr/AcOH, but not TFA. Their response to acidic media is thus similar to the Z group (also a benzyl ester).
t-Butyl Esters
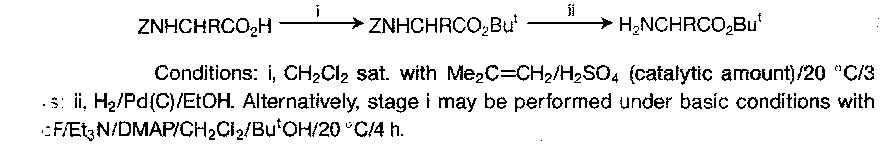
These are stable in the free base for (t-butyl ester inert to nucleophilic and basic attack). The stability and lability of these generally parallels the Boc group, as would be expected, so TFA will cleave it.
Phenyl Esters
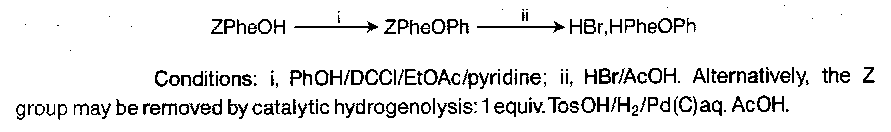
The group is incompatible with standard coupling and isolation routines. It is completely stable to acidic media (hence its development) as well as catalytic hydrogenolysis. Alkali causes cleavage more rapidly than with methyl esters, but it is the extraordinary susceptibility to attack by the peroxide ion which makes the method attractive. Mechanism presumably generates the peracid, but this is unstable with the α-electron withdrawing group present, so it collapses to the desired carboxylic acid.
Phenacyl Esters
-CO2CH2COPh. Formation by reaction of carboxylate with phenacyl bromide, BrCH2COPh. Stable in dry acidic conditions, and removable by zinc-acetic acid (orthogonal to t-Bu and Benzyl based protecting groups).
Peptide Bond Formation
With very few exceptions, peptide bond forming procedures involving the generation and aminolysis of reactive carboxy derivatives are employed. Activation, i.e. the attachment of a leaving group to the acyl carbon of the carboxy component, is necessary because carboxylic acids form salts with amines at ambient temperature.
There are three different ways of coupling along these lines:
- Those in which a reactive acylating agent is formed from the carboxy component in a separate step or steps, followed by treatment with the amino component,
- Those in which an isolable acylating agent is formed separately, and may be purified before aminolysis, and
- Those in which the acylating intermediate is generated in the presence of the amino component, by the addition of an activating agent to a mixture of the two components.
The activation is usually achieved by reaction of the carboxy component with an electrophilic reagent, either by adding:
Or by substitution:
Activation and Coupling
Acyl Chlorides and Fluorides
Conversion of acylamino acids to acyl chlorides, followed by reaction with amino acids or esters under Schotten-Baumann conditions is an obvious approach.
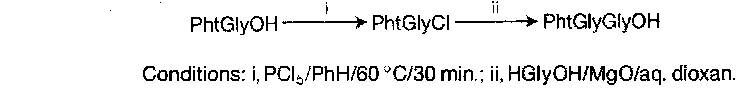
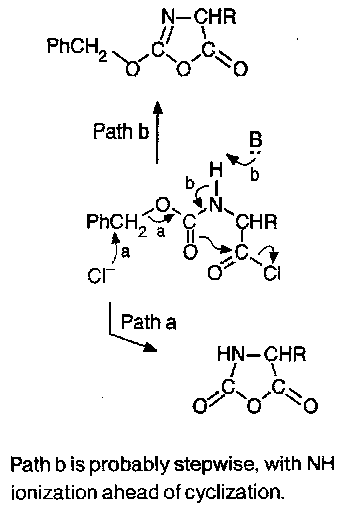
The reagents traditionally used for formation of the acyl chloride are thionyl chloride, PCl5, or similar. These are, however, too vigorous to be compatible with complex or sensitive substrates, and there is the risk of cyclisation to give oxazolones and hence racemic peptides.
Z amino acid chlorides are isolable but unstable, decomposing to N-carboxyanhydrides on warming (path a), although they cyclise to give benzyloxyoxazolones under basic conditions (path b). This position was altered dramatically by the discovery of Fmoc amino acid chlorides and also fluorides are easy to prepare, and are rather stable convenient intermediates.
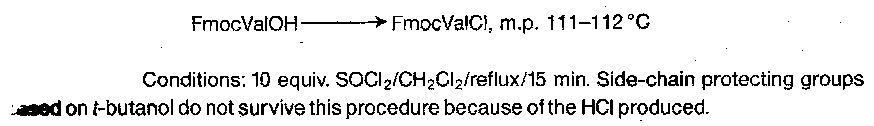
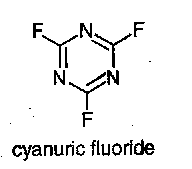
Fmoc amino acid fluorides can be obtained from Fmoc amino acid pyridine salts by treatment with cyanuric fluoride, a procedure which does not disturb t-butyl type protecting groups. Acyl fluorides are more stable than the corresponding acyl chlorides to neutral oxygen nucleophiles such as water, and to tertiary bases, but they show similar high reactivity to amino groups. They are of particular value for forming peptide bonds between sterically hindered amino acids such as α-aminoisobutyric acid (Aib).
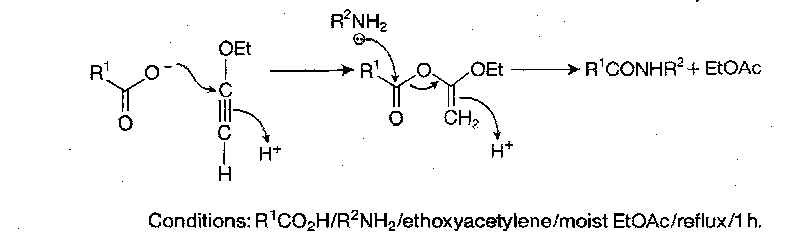
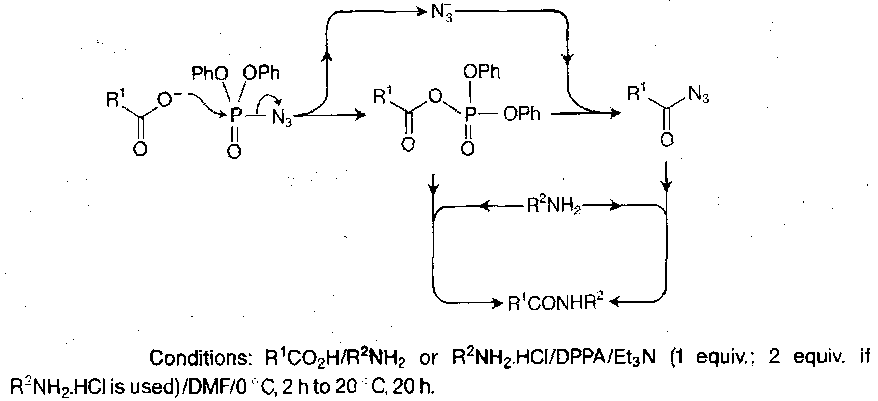
Acyl Azides
Made from hydrazides, which are obtained by hydrazinolysis of a protected amino acid or peptide ester, or by selective deprotection of a peptide derivative which has been constructed using a blocked hydrazide for carboxy-terminal protection. Conversion to the azide can be achieved by adding sodium nitrite in HCl(aq), but dry acid + nitrite ester in a dry organic solvent at lower temperature is now preferred.
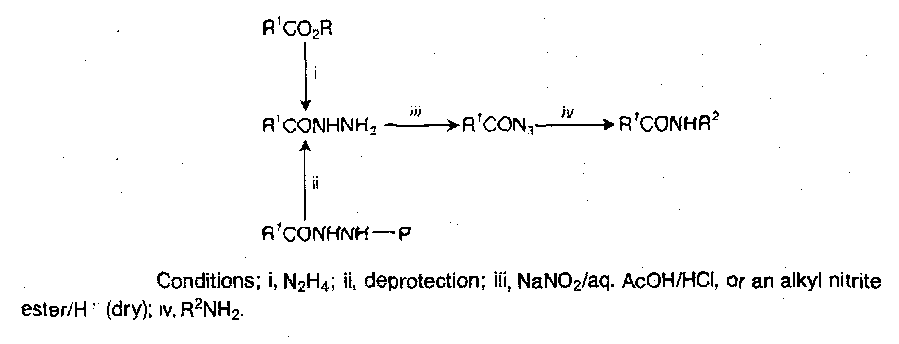
There are numerous side reactions possible with this procedure though (e.g. Curtius rearrangement to RNCO), so the azide method has not been used routinely for stepwise peptide building for a while. It remained a technique for segment condensation, as the procedure is free from racemisation. Racemisation only ever occurs with excess base in polar solvents, but it remains the least likely method for this to occur.
Anhydrides
Symmetrical Anhydrides
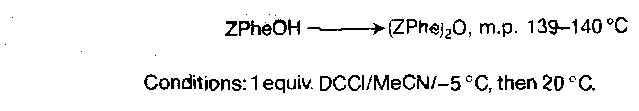
Aminolysis of these is unambiguous, but only half the carboxy component is incorporated into the product. They are otherwise clean acylating agents, with the exception of Z-glycine derivatives being unstable on heating or with base. They are very useful in stepwise synthesis.
Mixed Anhydrides with Carboxylic Acids
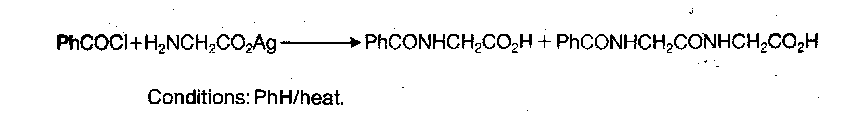
Main problems with this method are that the anhydride has two similar electrophilic sites and can therefore undergo aminolysis ambiguously, and also these reagents tend to disproportionate, ultimately with the same result.
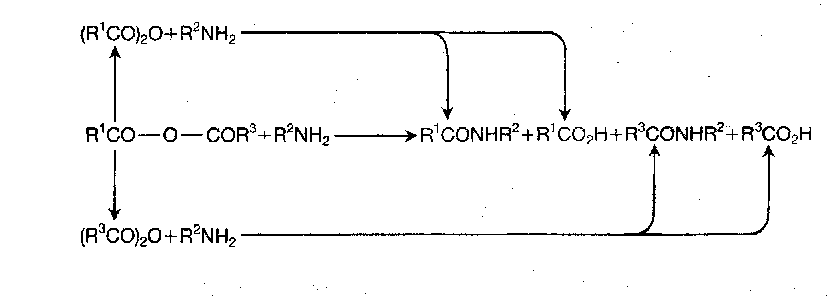
However, the selectivity can be improved greatly by introducing steric hindrance and inductive depression of electrophilicity, so as to direct the attack to the carboxy component carbonyl, as in mixed pivalic anhydrides:
Mixed Pivalic Anhydrides are commonly used as they are stable wrt disproportionation, and can be isolated in some cases. Pivaloyl derivatives of the amino component are occasionally obtained, but this is not normally a serious matter, and the method is a valuable alternative to the mixed carbonic anhydride method.
Mixed Anhydrides with Carbonic Acids
This is the most popular of the anhydride methods.
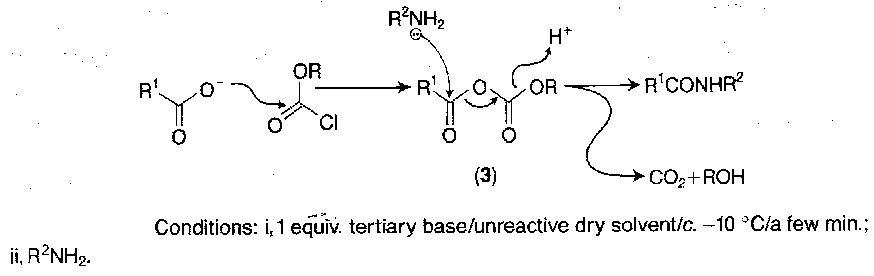
Ethyl chloroformate is commonly used, but isobutyl chloroformate is also popular. The flanking of one of the carbonyl groups by two oxygen atoms diminishes reactivity, so that nucleophilic attack is directed towards the carbonyl of the original carboxy component.
Low temperatures and minimal activation times are usually employed:
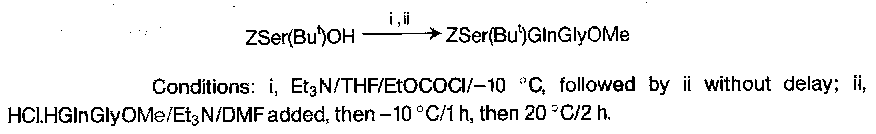
They can be isolated, although there is some decomposition at ambient temperature. The timescale for these reactions is measured in hours rather than minutes. Conditions for the minimisation of racemisation have been delineated for model cases, but the technique has not found wide application for fragment condensation at racemisable residues, as less risky procedures are available.
The value of the method lies in its speed and economy, and it has been used in repetitive stepwise synthesis with some success, up to medium chain length. Its most common use is in simple protected peptides in the tri- to hexapeptide range, as these have solubility properties uncomfortably similar to those of dicyclohexylurea (making DCCI methods tiresome).
To avoid the need for separate activation and aminolysis steps, mixed ethyl carbonic anhydrides can be generated by the reaction below of carboxylic acids with 1-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline (EEDQ):
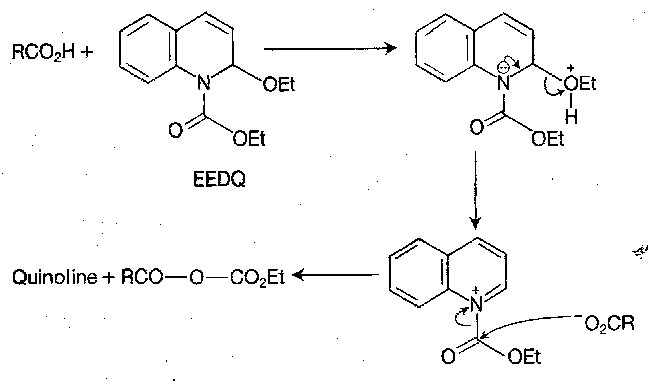
EEDQ can also be used as a direct coupling reagent:
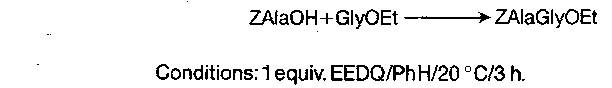
Because in this technique the mixed carbonic anhydride is consumed by aminolysis as soon as it is formed, the opportunity for the intervention of side-reactions, including racemisation, is minimal. Workup is also simple.
Mixed Anhydrides with diphenylphosphinic acid
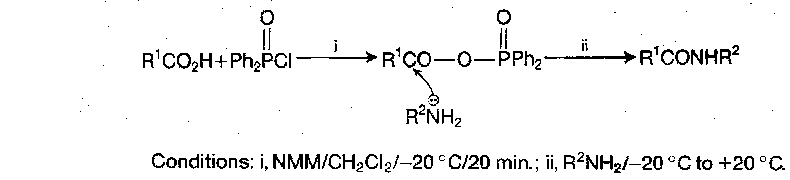
This method is more regioselective than the other mixed anhydrides, as aminolysis occurs exclusively at the carbonyl of the activated amino acid residue, even when steric factors might militate otherwise. Coupling is thus very clean and workup is simple.
Carbodiimide Reagents
Dicyclohexylcarbodiimide (DCCI) has been the single most important reagent for activating carboxy groups in peptide synthesis ever since it was reported in 1955. It can be used to generate activated carboxy derivatives such as symmetrical anhydrides (as earlier) and active esters (see later), or as a direct coupling reagent. In all cases, the primary activating event is addition of the carboxy group to the carbodiimide functionality to give an O-acylisourea, which is a potent acylating agent.
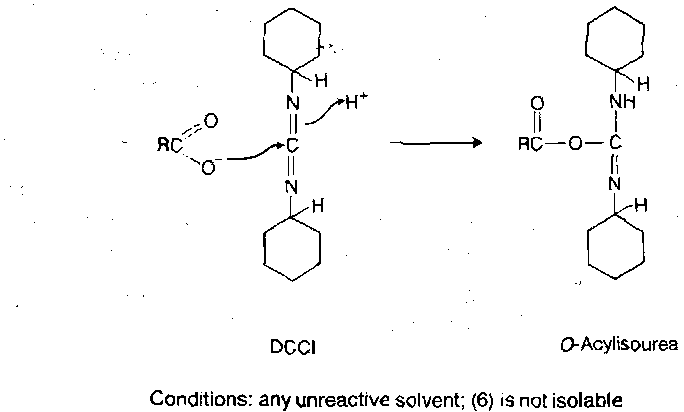
Direct peptide coupling with DCCI is in principle very simple, involving mere mixing of the amino and carboxy components with DCCI in equimolecular amounts in an organic solvent at ambient temperature. O-acylisourea formation is rapid, leading to peptide either by immediate aminolysis or via a symmetryical anhydride, with the concomitant formation of dicylcohexylurea.
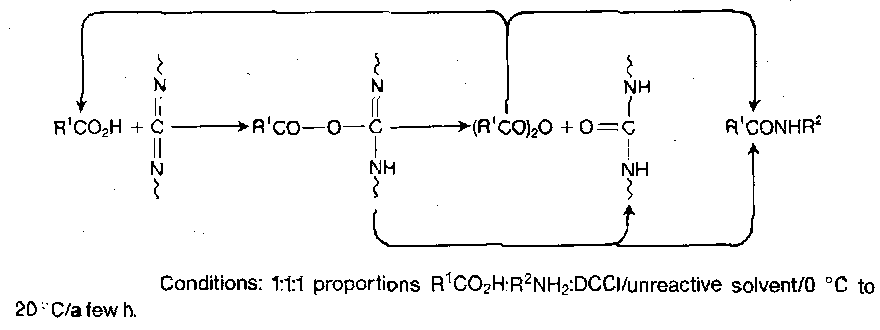
The urea is only sparingly soluble in most solvents, so its separation is facile in solution synthesis, while being soluble enough to remove through washing in solid phase synthesis.
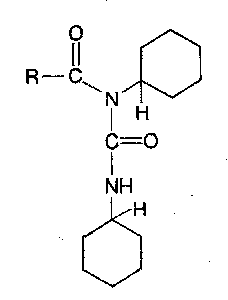
However, the intermediates shown are highly reactive, and side reactions can intervene, especially if the amino component is dilatory. Extensive racemisation takes place with susceptible carboxy components. Furthermore, the collapse of the O-acylisourea by intramolecular acyl transfer sometimes competes significantly with the desired attack by external nucleophiles. When this happens, the much less reactive N-acylurea below is formed. This can also be formed by intermolecular reaction between the urea and a symmetrical anhydride, especially in DMF.
N-acylurea formation not only reduces the yield, but may give rise to purification problems. This difficulty, and the danger of racemisation, can both be greatly reduced by performing the coupling in the presence of a suitable α-nucleophile which is able to react very rapidly with the O-acylisourea before side-reactions can intervene. An acylating agent of lower potency is formed, which is still reactive with respect to aminolysis, but which is more discriminating and does not lead to racemisation or other side-reactions.
Numerous possible additives, which in effect generate an active ester in situ, are used. N-hydroxysuccinimide was the first, but 1-hydroxybenzotriazole (HOBt) has been the one most regularly used so far. The recently introduced aza and monocyclic analogues HOAt and HOCt may prove to be superior.
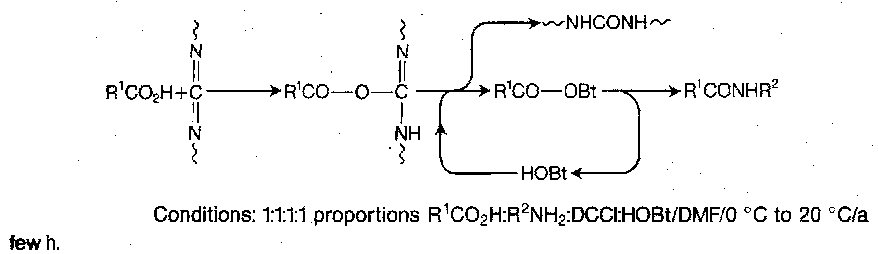
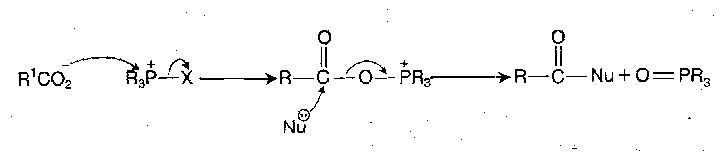
Phosphonium and (so-called) Uronium Reagents
Acyloxyphosphonium species react readily with nucleophiles at the acyl carbon.
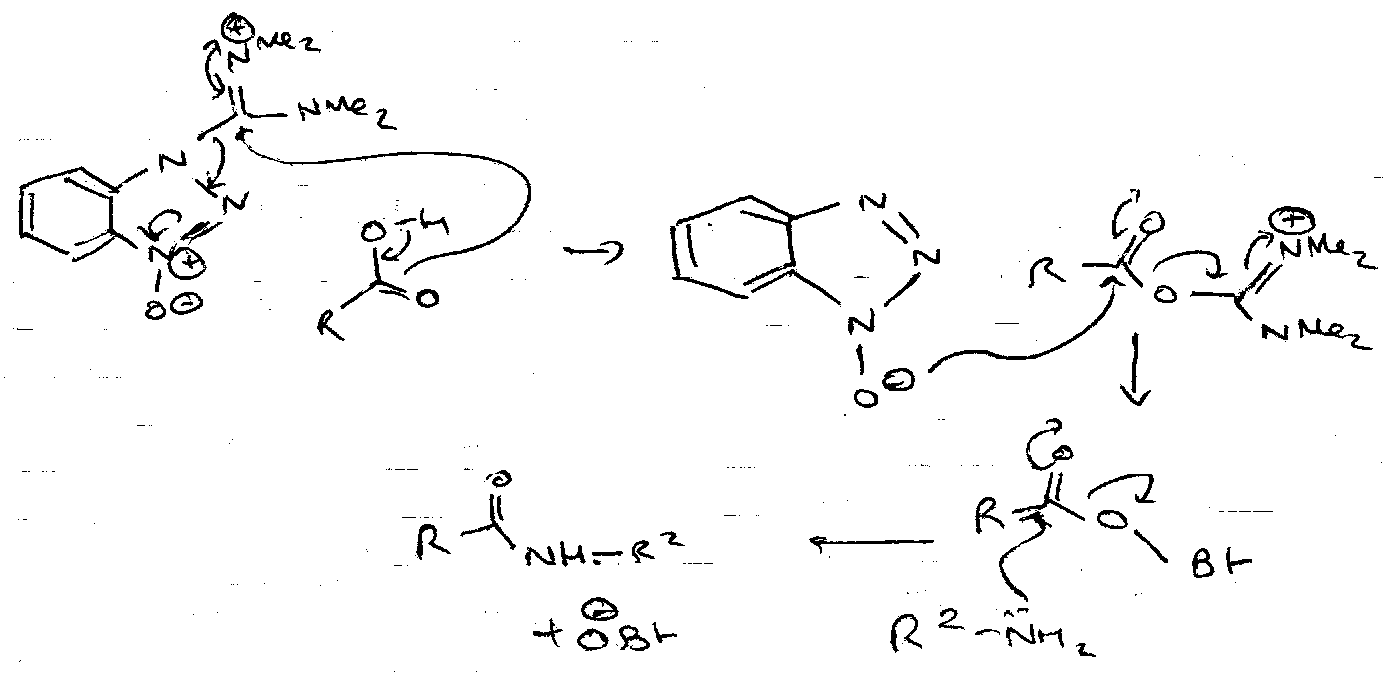
Benzotriazolyloxy-tris-(dimethylamino) phosphonium hexafluorophosphate, alias the BOP reagent, is the main one. It has the formula (Me2N)3P+--OBt,PF6-. For coupling, an equivalent of the reagent is added to a 1:1 mixture of the amino and carboxy components, together with a tertiary base to ensure that the latter is ionic. Many pathways are possible, but the main one is via the ester of HOBt:
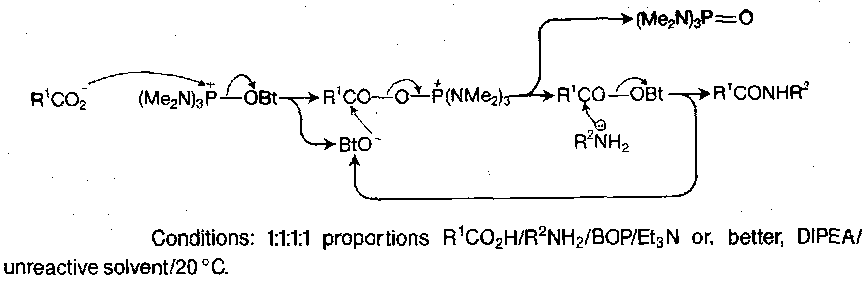
All the co-products are easily removed, and the procedure gives high yields with few side-reactions, even in difficult cases. As the initial step is anion-cation reaction, it is favoured in less polar solvent. It is also applicable in both solution and solid phase.
New reagents termed TBTU and HATU are replacing this on grounds of toxicity.
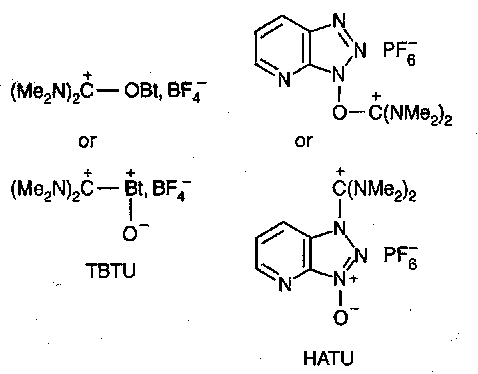
These are obtained from HOBt / HOAt respectively, by reacting them with tetramethylchloroformadinium cation (Me2N)2C+--Cl (made from phosgene or oxalyl chloride on tetramethylurea).
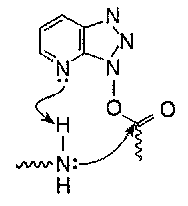
TBTU and HATU are termed uronium salts, despite the fact that they are actually guanidinium derivatives in the solid state. In solution the two forms may coexist. HATU seems to be the best reagent in this class, succeeding in sterically hindered couplings and giving minimal racemisation. The high reactivity to amino groups is probably due to intramolecular general base catalysis.
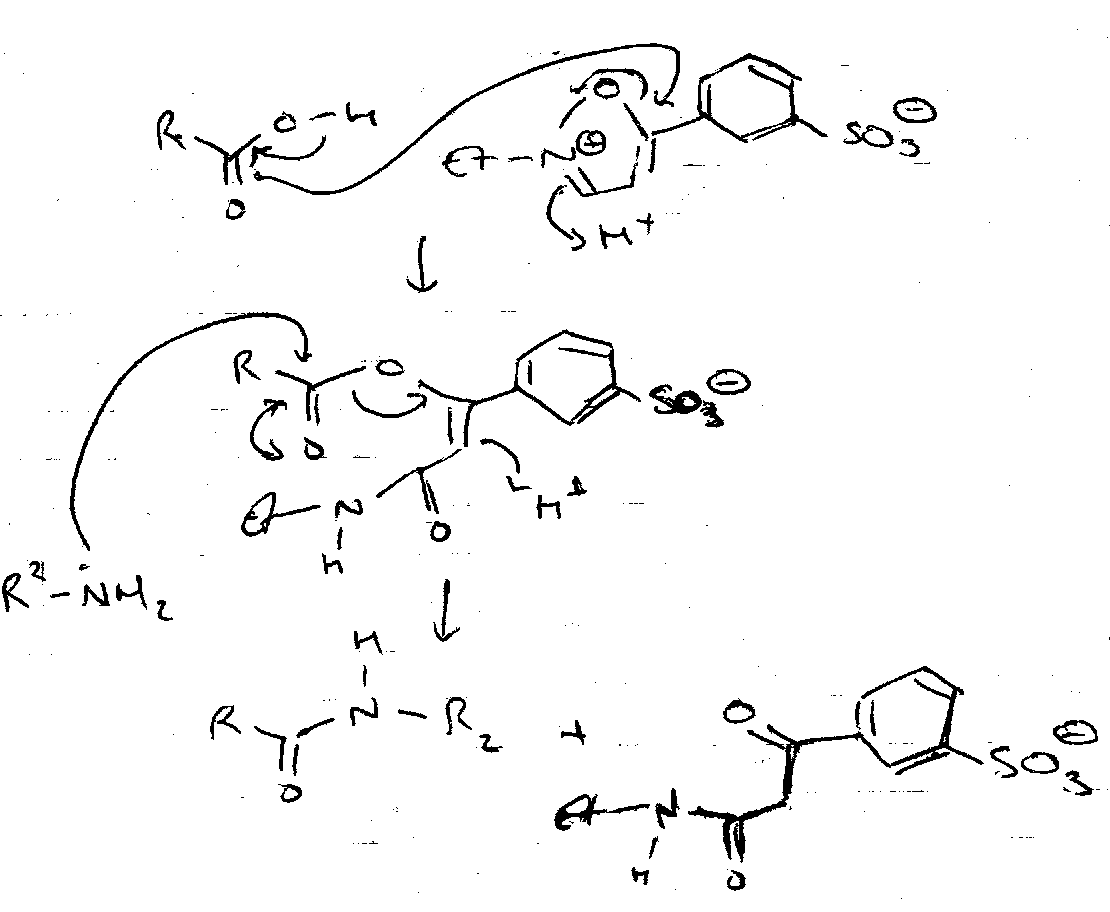
Mechanism –
Note that another Direct Coupling Reagent, Woodward’s Reagent K, is also often used:
Active Esters
Active esters offer excellent leaving groups attached to the carbonyl, for rapid aminolysis. They are usually prepared by DCCI-mediated coupling:
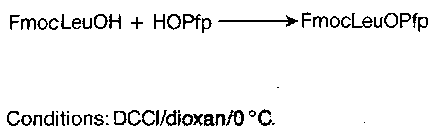
Mixed anhydride methods can also be used, and various specialised reagents are available:
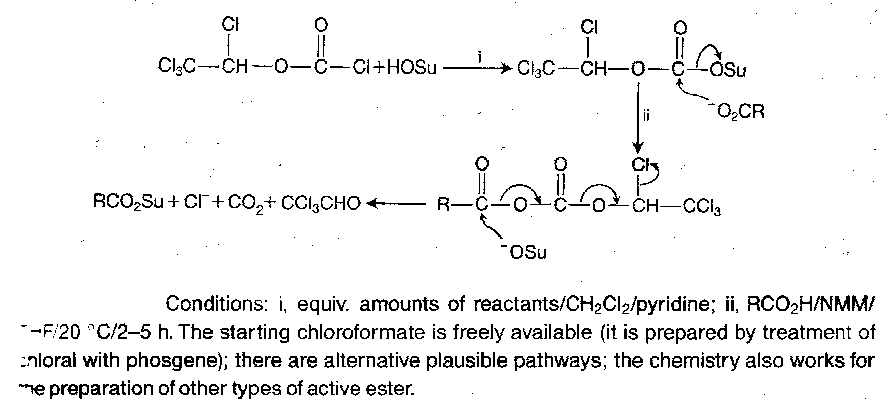
The currently popular active esters are generally crystalline, stable materials.
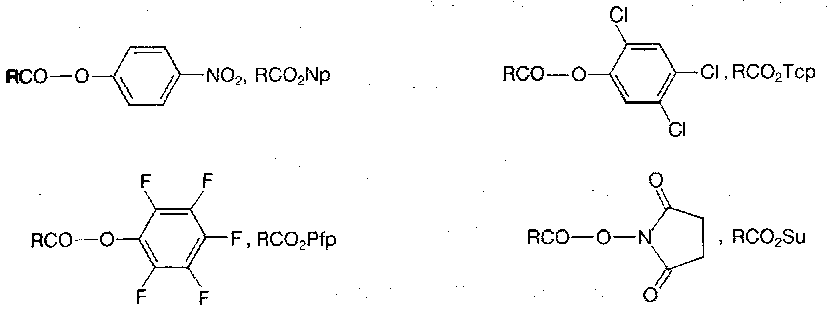
Because they are at a low level of activation, side-reactions during coupling (including racemisation) are generally less of a problem than with most peptide bond forming procedures, although there is a serious risk of racemisation when active esters are prepared directly if the carboxy component is susceptible, especially if exposure to base is involved.
The appeal lies in the cleanliness of the reaction, allowing easy repetition which dispense with isolation of intermediate peptides. The more reactive active esters are also valuable in solid phase synthesis.
The reactivities of the various active esters correlates broadly with the acidities of the ester moieties, after allowance for sterics etc. Pfp and Su are especially reactive. Co-product removal is also a consideration.
N-carboxyanhydrides
The N-carboxy anhydrides (NCAs) of α-amino acids are easily obtained by the action of phosgene of α-amino acids, or by thermal decomposition of alkoxycarbonyl-α-amino acid chlorides. They are useful in two modes.
First, treatment with small amounts of nucleophilic initiator in an organic solvent leads to successive ring opening and loss of carbon dioxide, giving a new nucleophile which attacks another molecule of NCA, and so on, leading to the formation of homopolyamino acid:

Second, providing the conditions are carefully controlled, NCAs can be used for rapid peptide synthesis in aqueous solution:
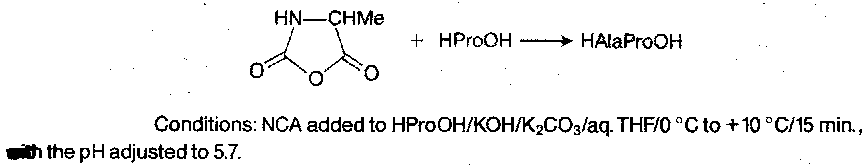
Extension to short oligopeptides is possible by repetition, e.g.
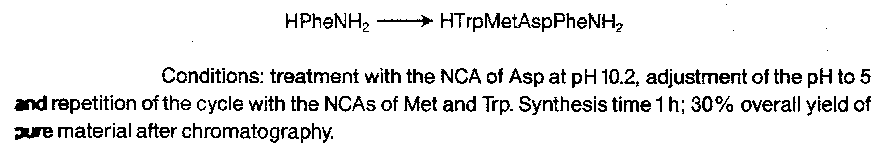
The peptides prepared via NCAs in this way are of high optical purity, and in principle the great rapidity of the technique and the possibility of using a repetitive regime makes it very attractive.
However, the carbamic acids produced by aminolysis of NCAs are unstable, and some decarboxylation usually occurs before the aminolysis stage is complete, giving a fresh amino group which can react with any remaining NCA and result in “over-reaction”. Minimisation of this and other side-reactions requires exceedingly careful control of the reaction conditions, and the progressive accumulation of over-reaction products limits the extent to which the repetitive approach can be taken without prohibitive purification problems.
NCAs can be N-alkoxycarbonylated in the presence of base to give urethane-protected NCAs (UNCAs). Fmoc-NCAs, for example, are generally useful and can be kept for some time, and react rapidly and cleanly with amino groups to give Fmoc-aminoacyl derivatives and carbon dioxide as the only co-product:
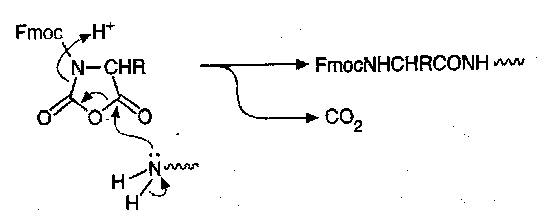
Racemisation
In peptide chemistry this term covers not only conversion of enantiomers but also partial epimerisation, whereby there is loss of chiral integrity at one of two or more chiral centres, resulting in the formation of a mixture of epimers (i.e. diastereoisomers differing at one chiral centre).
It is an almost exclusively base-induced side-reaction, and in practice is only a matter for serious concern at the activation and coupling stages of a synthesis. There are two principal mechanisms.
Direct Enolisation
Deprotonation at the α-carbon of an α-amino acid residue result in racemisation, because the carbanion can reprotonate on either side:
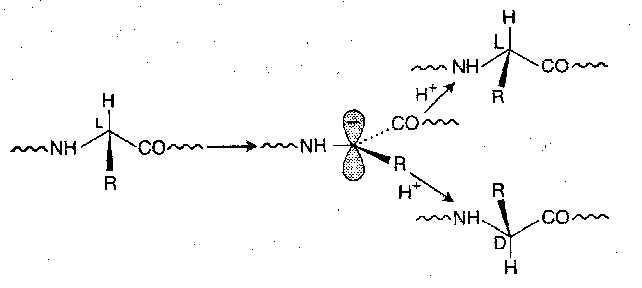
When the group attached to the carbonyl is NH, O-alkyl or O-, it is in most cases negligible, so deprotection is safe. During activation and coupling, however, the risk is more significant. Racemisation is fastest when electron-withdrawing groups are in the position next to the carbonyl (i.e. good leaving groups) and with unhindered strong bases in dipolar aprotic solvents like DMSO and DMF. Fortunately, with the exception of a few special amino acids and couplings which are extremely slow, the amount of racemisation which takes place is very slight.
The Oxazolone Mechanism
Activated acylamino acids and peptides cyclise under the influence of base to give oxazolones. These are themselves activated towards aminolysis, and reaction with amino components leads ultimately to peptides, but since their racemisation via stabilised anions is usually fast compared to the rate of peptide bond formation, any peptide produced is largely useless.
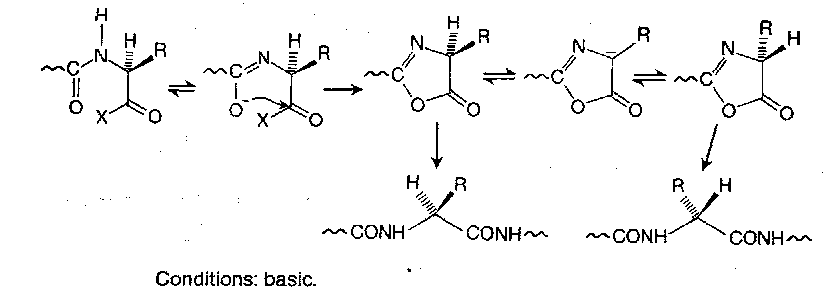
When the amino-nitrogen of the activated residue is acylated with a simple acyl group (acetyl, benzoyl, etc), or with a peptide chain, cyclisation to the oxazolone occurs easily with most good leaving groups X, and gross or even complete racemisation may ensue. But oxazolone formation is not so facile when the acyl substituent is an alkoxycarbonyl protecting group. This is vitally important, and explains the extensive use of alkoxycarbonyl protection.
Use of Enzymes
Advantages here are the mild conditions, freedom from racemisation and the need for side-chain protection, and scope for industrial scale-up.
Disadvantages – unpredictability. Also, not all the appropriate proteases are available, and non-proteinogenic amino acids are obviously problematic.
Residue-Specific Considerations
Lysine
Block the ε-amino group on the side-chain prior to peptide bond formation. Can be achieved using the fact that the side-chain amino group is the more basic and nucleophilic because there is an electron-withdrawing group next to the α-amino group, or by taking advantage of the fact that two α-functionalities can engage in chelate formation.
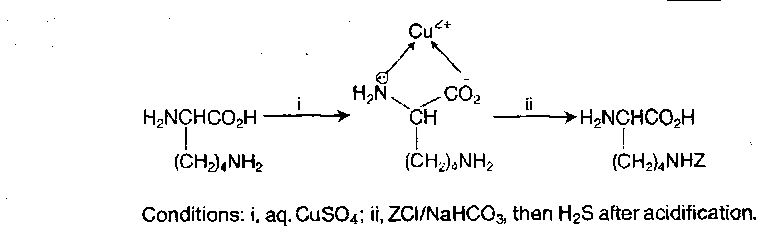
Once the amino groups have been distinguished, trivial reactions give ready access to any required orthogonally disubstituted lysines, e.g.
Arginine
Guanidino side-chain is a strong base which remains protonated under normal conditions for α-protection, peptide bond formation, and deprotection. Thus, it can be left unprotected. However, may give rise to solubility problems, so protection is often carried out. The classic method is nitration:

Note that there are risks of side-reactions in base (cyclisation).
Aspartic and Glutamic Acids
Peptide bond formation involving α-carboxy groups requires that the carboxy groups on these side chains must be protected (obviously!). Benzyl or t-butyl ester protection is usually employed.
Differentiation of the carboxy groups is again based on reactivity – the α-carboxy is deactivated to esterification by a nearby protonated amino group.
Main difficulties lie in the ease of cyclic imide and amide ring formation, and may be one of the most serious side-reactions in peptide synthesis:
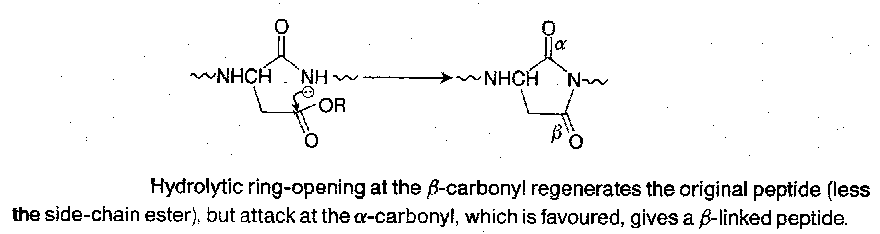
It is less serious with secondary and tertiary alkyl esters in the side-chain than it is with esters of primary alcohols. 5-membered ring cyclisation is more rapid, so it is less of a problem for glutamic acid.
Cysteine and Cystine
Unique case, due to disulphide bridge formation. Until we want these to form, the thiol groups must be protected, because they interfere with many routine operations. They are soft nucleophiles, so selective S-protection by direct alkylation is easy.
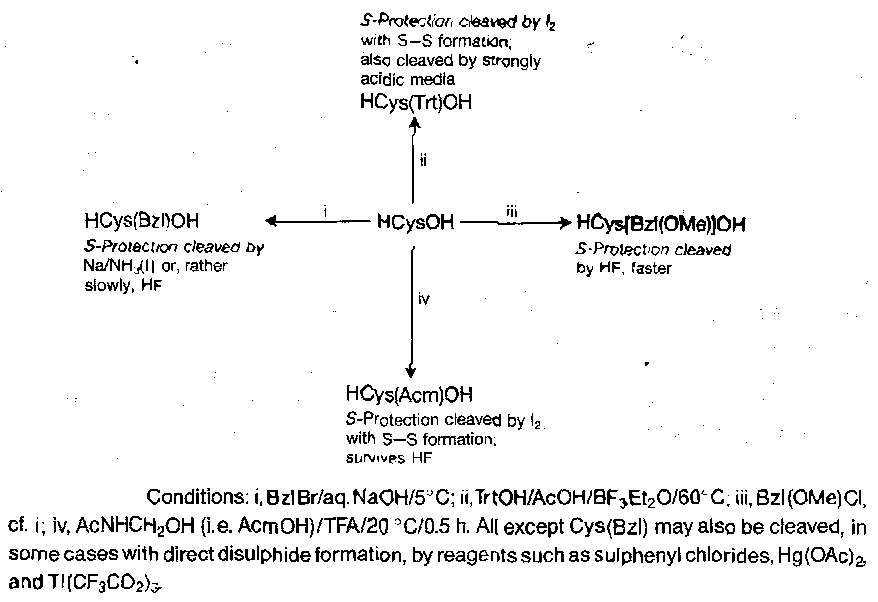
None of the S-protecting groups are compatible with ordinary catalytic hydrogenolysis, but H2/Pd in liquid ammonia may be used for removing Z from Cys(Bzl) derivatives. Mild acid and base α-deprotective procedures may be used in conjunction.
Forming disulphide bridges can be carried out by oxidation of unprotected thiols, or more usually now without deprotection, by iodine oxidation of bis-trityl peptides.
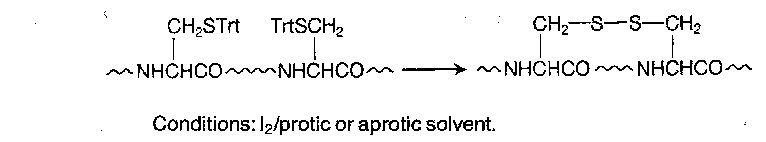
The reaction probably proceeds as follows:
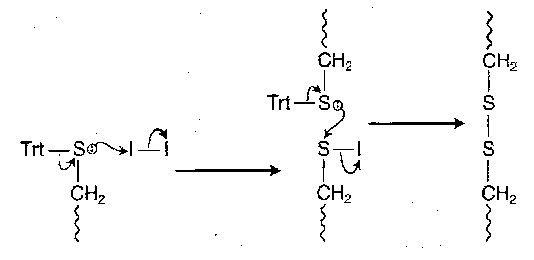
For more than one disulphide bridge in a molecular, fragmentation tactics must be used, or selective deprotection of the appropriate thiols in turn.
Methionine
This is problematic because the thioether side-chain interferes with catalytic hydrogenation, and is liable to be attacked by electrophiles generated on acidolytic deprotection of other functionalities. The usual response is, however, to live with it, adjusting the conditions chosen accordingly. Protection by reversible oxidation to the sulphoxide with H2O2 is favoured by some.
Serine, Threonine, Tyrosine
Hydroxy groups, especially phenolic ones, react with acylating agents, and are therefore usually protected in peptide synthesis. They can sometimes be left without in conditions of mild carboxy-activation. Benzyl ether protection, cleaved by vigorous acidolysis or catalytic hydrogenolysis, or t-butyl ether protection (TFA cleavage) are standard.
Asparagine and Glutamine
Amide side-chains are usually left unprotected, despite poor solubility and risk of side-reactions. Trityl protection is sometimes used in conjunction with Fmoc α-protection, as it is easily removed by mild acid at the end, but resists catalytic hydrogenolysis and base.
Histidine
The basic and nucleophilic side-chain requires protection. Racemisation is particularly prevalent, as is attack on DCCI, and formation of acyl-imidazoles.
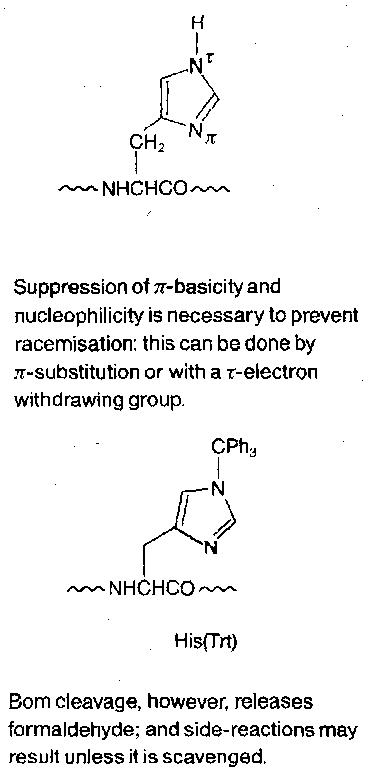
As shown, the trityl group is currently the protecting group of choice, placed at the τ-position. Racemisation is still a problem due to the free π-position. In conjunction with Boc α-protection, π-benzyloxymethyl (Bom) protection is ideal – it resists TFA, but is cleaved by HF or HBr/AcOH and (less reliably) by catalytic hydrogenolysis.
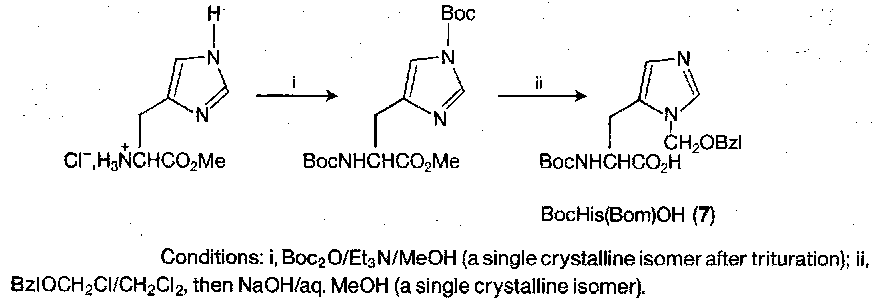
Tryptophan
The indole ring does not interfere with peptide bond formation, but captures electrophiles with great ease at several sites, especially the 2-position, meaning scavengers for deprotection of acid labile protecting groups are essential.
Proline
The special structure of proline gives it two impediments in synthesis (steric hindrance to acylation, susceptibility of Xaa-Pro bonds to reduction by sodium in ammonia), but also gives it a resistance to base-catalysed racemisation which is unique among the chiral protein building blocks. From a strategic point of view, this is a vital fact, because it allows any coupling from a proline carboxy group, including an acylpeptide fragment, to be performed without racemisation.
The ordinary oxazolone racemisation mechanism pathway is not possible because they have no NH. Cyclisation to an oxazolonium cation can take place, and such cations are optically labile, but much more vigorous conditions are required than are ever used in peptide synthesis.
Glycine, Phenylalanine, Alanine, Leucine, Isoleucine, Valine
No side chain functionality. Alanine and Leucine are trouble-free.
Glycine, achiral and without any side chain, is sometimes difficult to deal with, because the unimpeded access to peptide bonds involving it allows side reactions such as hydantoin formation. The same consideration facilitates strong interchain H-bonding, which tends to make peptides with a high glycine content rather insoluble.
Phenylalanine is only a problem if catalytic hydrogenolysis reaction times are too long (is reduced to cyclohexylalanine).
Valine and Isoleucine couple slowly (steric reasons) – allows side-reactions to compete.
Strategy and Tactics
Stepwise Elongation Strategy – e.g. for a tripeptide, [1+(2+3)] is the same as [(1+2)+3].
For a tetrapeptide, however, there are five strategies. Four of these are formally stepwise, but one is convergent, i.e. two dipeptides are connected together. This has implications for yield. Assuming there is an 80% yield at each stage, then stepwise elongation gives a total yield of 52%, while convergent (fragment) methods gives 64%.
There are also further advantages to the fragment condensation approach. Firstly, if small fragments are combined to make one which is larger, the differences between the desired product and everything else ought to facilitate its isolation, whereas the addition of residues one at a time would result only in progressive small changes in chemical character and molecular size, with consequently greater isolation and purification problems. Secondly, in a fragment condensation approach, personnel can be assigned each of their fragments to work simultaneously in parallel.
However, there are contrary considerations. Fragment condensation is better performed at some positions than others, so it is a constrained strategy, and its protective group tactics are in general more complicated than those of a stepwise synthesis strategy from the carboxy end.
For a classical solution synthesis, the optimum approach is usually stepwise synthesis of fragments for assembly in an overall convergent strategy.
The principal strategic constraint in the desirability of proceeding in such a way as to minimise the risk of racemisation throughout. There is no real risk except at the activated amino acid residue during the activation and peptide bond formation stages. Furthermore, if the azide or DCCI/HOBt procedures are used, racemisation is usually slight, with small peptides at least. The situation is summarised more fully below:
|
A - Safe |
B - Usually Safe |
C - Unsafe |
|
Activation and coupling of any alkoxycarbonyl-amino acid by any conventional method |
Activation and coupling of any carboxy component by the azide method (unless coupling sluggish) |
Activation and coupling of any carboxy component not in column A by any method not in column B |
|
Activation and coupling of acyl-peptidylglycines and acylpeptidyl-prolines by any conventional method |
Activation and coupling of any carboxy component by the DCCI/HOBt method or a variant |
|
Advances in separation technology (e.g. countercurrent techniques) are taking place that minimise these risks further.
Generalising about synthesis strategies is usually not possible. The presence of disulphide bridges usually needs to be considered first. Problematic residues such as Met or Trp are preferably introduced late in the synthesis. It is also wise to consider whether fragments will end up being unreactive or insoluble.
The number and distribution of glycine and proline residues are important in the design of a fragment condensation strategy. This is because it prevents oxazolone formation when glycine is the C-terminus (see earlier). Also, proline locks the conformation (as does a disulphide bridge) preventing the cyclisation. For example, a fragment strategy for AlaGlyGluMetGluOH should be devised based on splitting between Gly and Glu, so that racemisation is avoided. Hence, Z-Ala-Gly-OMe and Boc-Glu(OBzl)-Met-Glu(OBzl)-OMe would be combined.
N to C strategies are usually highly undesirable. This is due to the fundamental flaw that 5-membered ring formation is favoured, i.e. oxazolone formation is a problem. Hence, carbamate protection is preferred as this prevents this happening, i.e. want C to N strategy.
Protecting Group Tactics
Consider the target GlyLysGly, which is best approached by the stepwise strategy:
The minimum requirement is that protecting group P2 is orthogonal to P1 and P3. P2 is termed the temporary protecting group.
The most important tactical approaches using well-tried protecting groups are shown:
|
Tactics |
A |
B |
C |
D |
|
Temporary α-amino |
Z |
Boc |
Bpoc |
Fmoc |
|
Cleaving temporary |
H2/cat |
Mild acid |
Very mild acid |
Basic |
|
Permanent protection |
Boc, OBut, etc |
Z, OBzl, etc. |
Boc, OBut, etc |
Boc, OBut, etc |
|
Cleaving permanent |
Mild acid |
Strong acid or H2/cat |
Mild acid |
Mild acid |
The above also represents a solid phase synthesis if P1 is the resin carrier, which can be regarded as the C-terminal permanent protecting group. In fact, B and D are the Merrifield and Sheppard methods (see later).
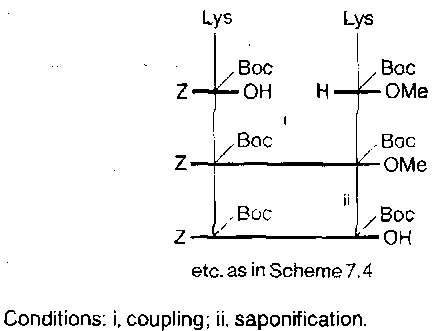
Tactics are more complicated for fragment condensation strategies. For example, Lys(Boc)Lys(Boc) insertion between two other fragments, to make –COLys(Boc)Lys(Boc)NH-, there are a number of options:
Salt coupling –

Methyl Ester –

Remember also that side chains (like Lys above) may need protecting. This was discussed earlier.
Solution Peptide Synthesis
Best described by examples.
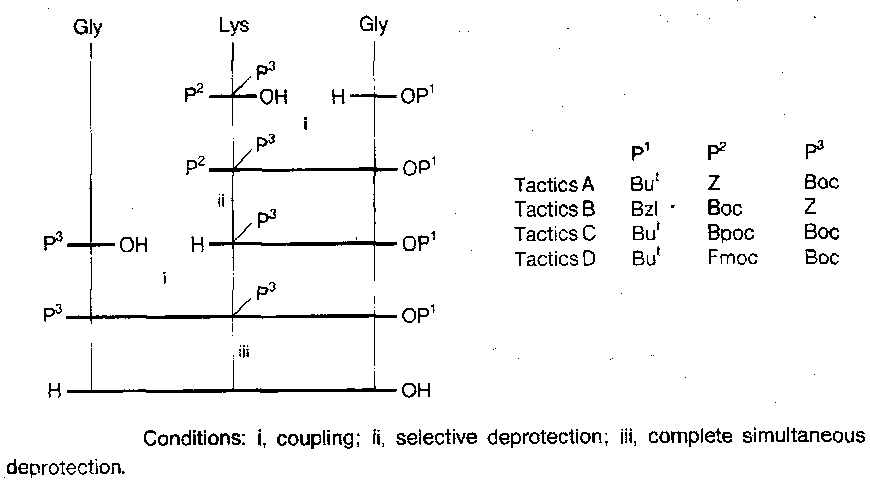
Oxytocin
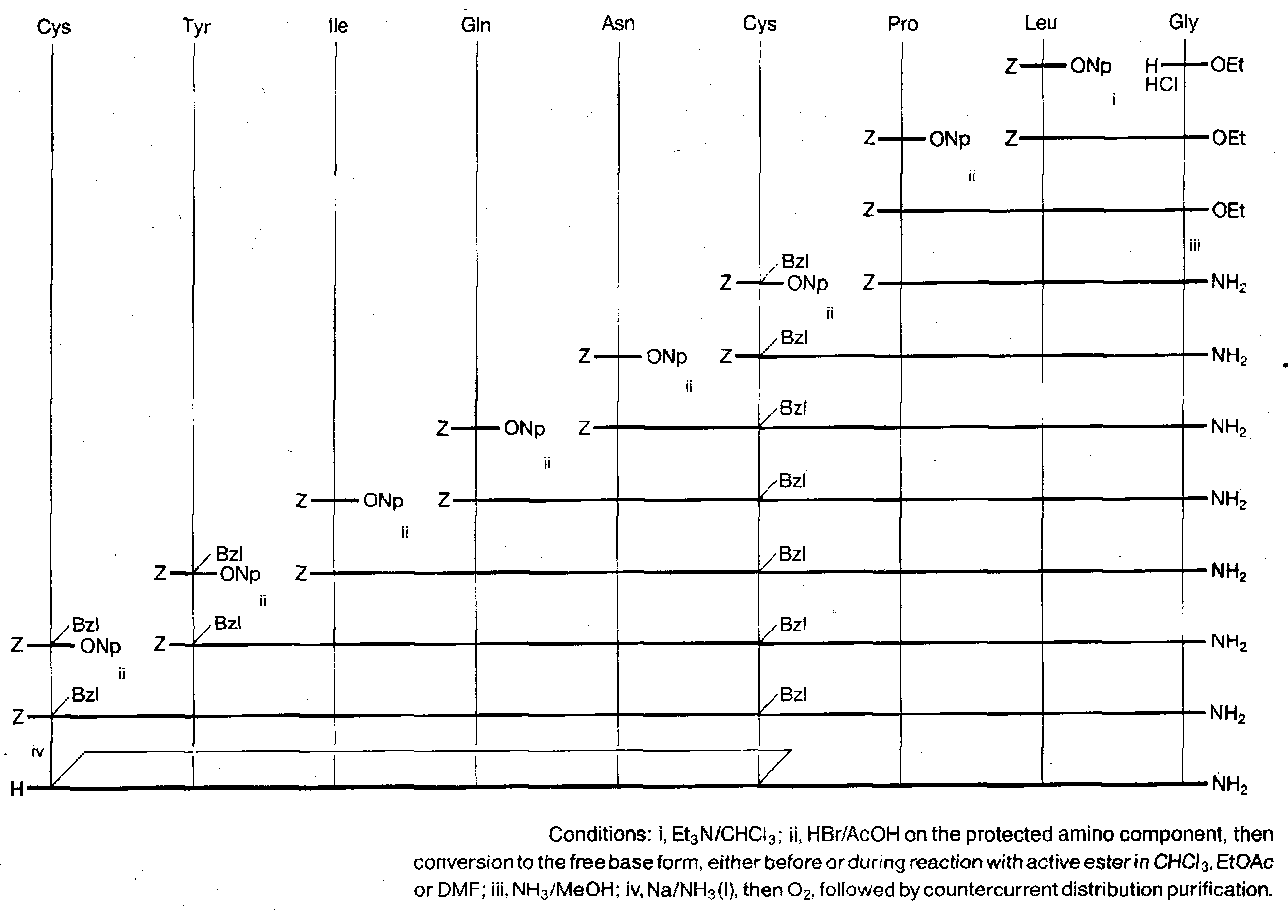
Also, an entirely different approach:
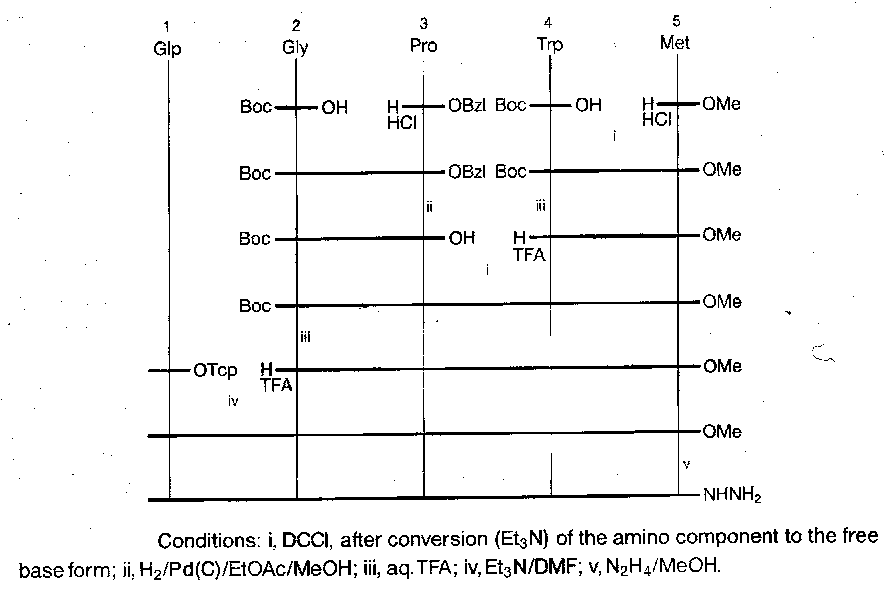
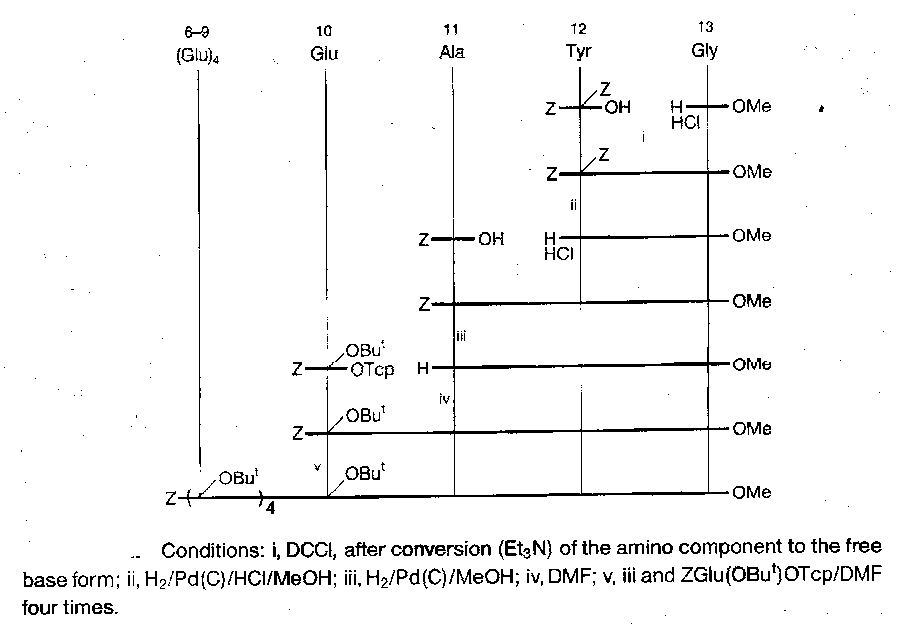
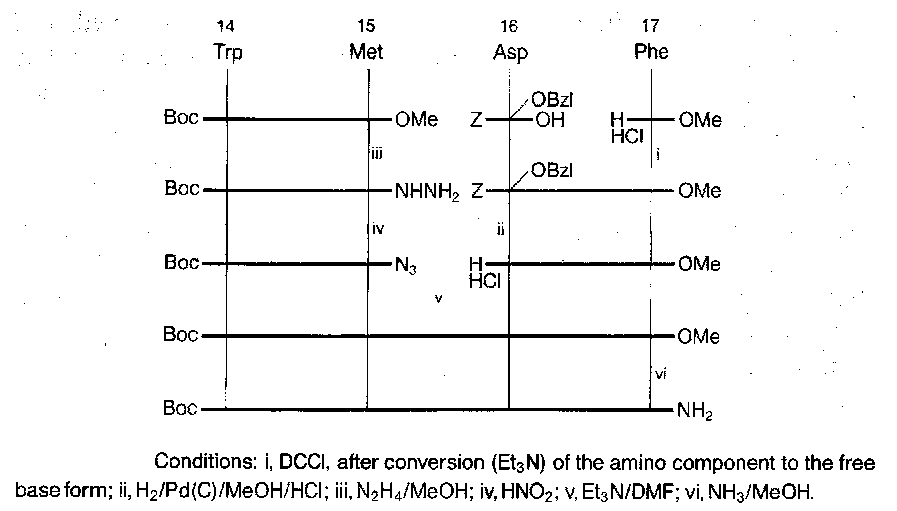
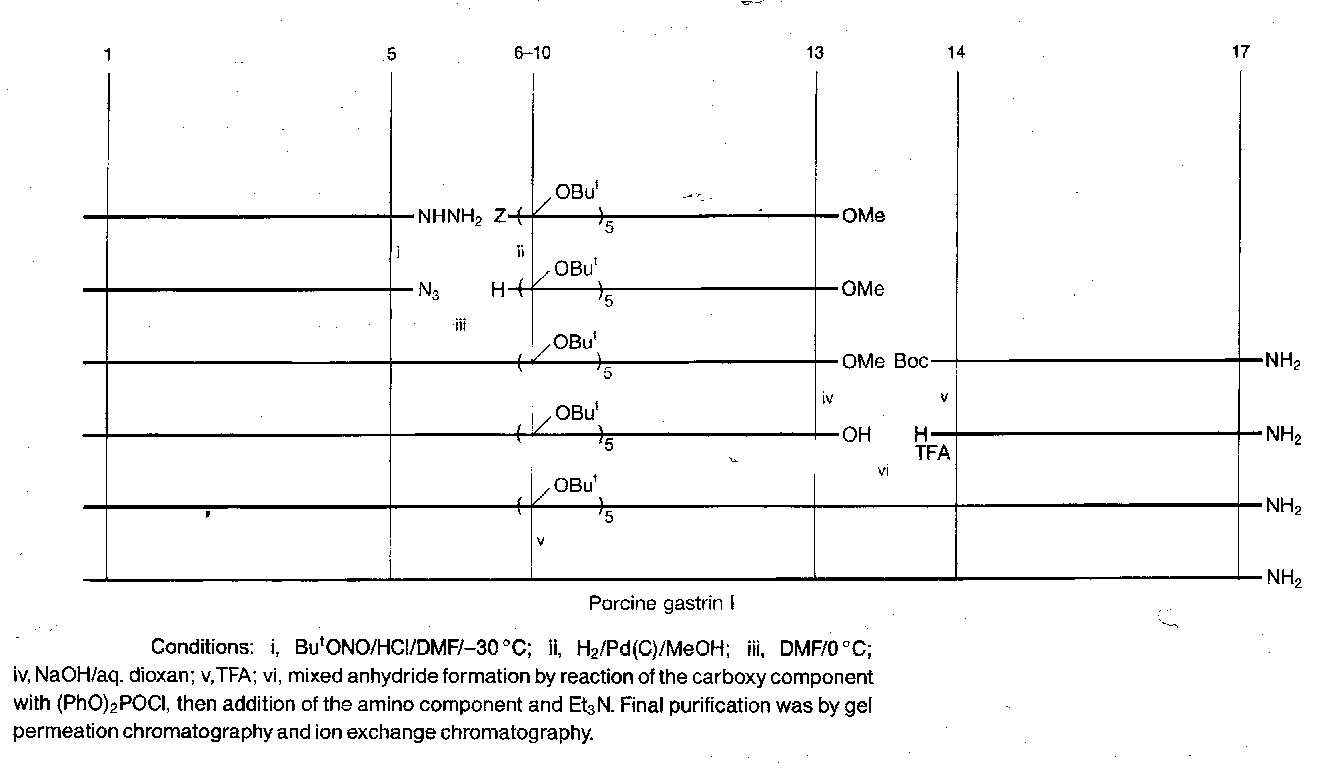
Porcine Gastrin I
Fragment condensation strategy adopted, with three main fragments of comparable size, which were chosen to exclude Met from the central fragment (allowing the use of hydrogenolysis). Fragment conjunction performed at points dictated by racemisation.
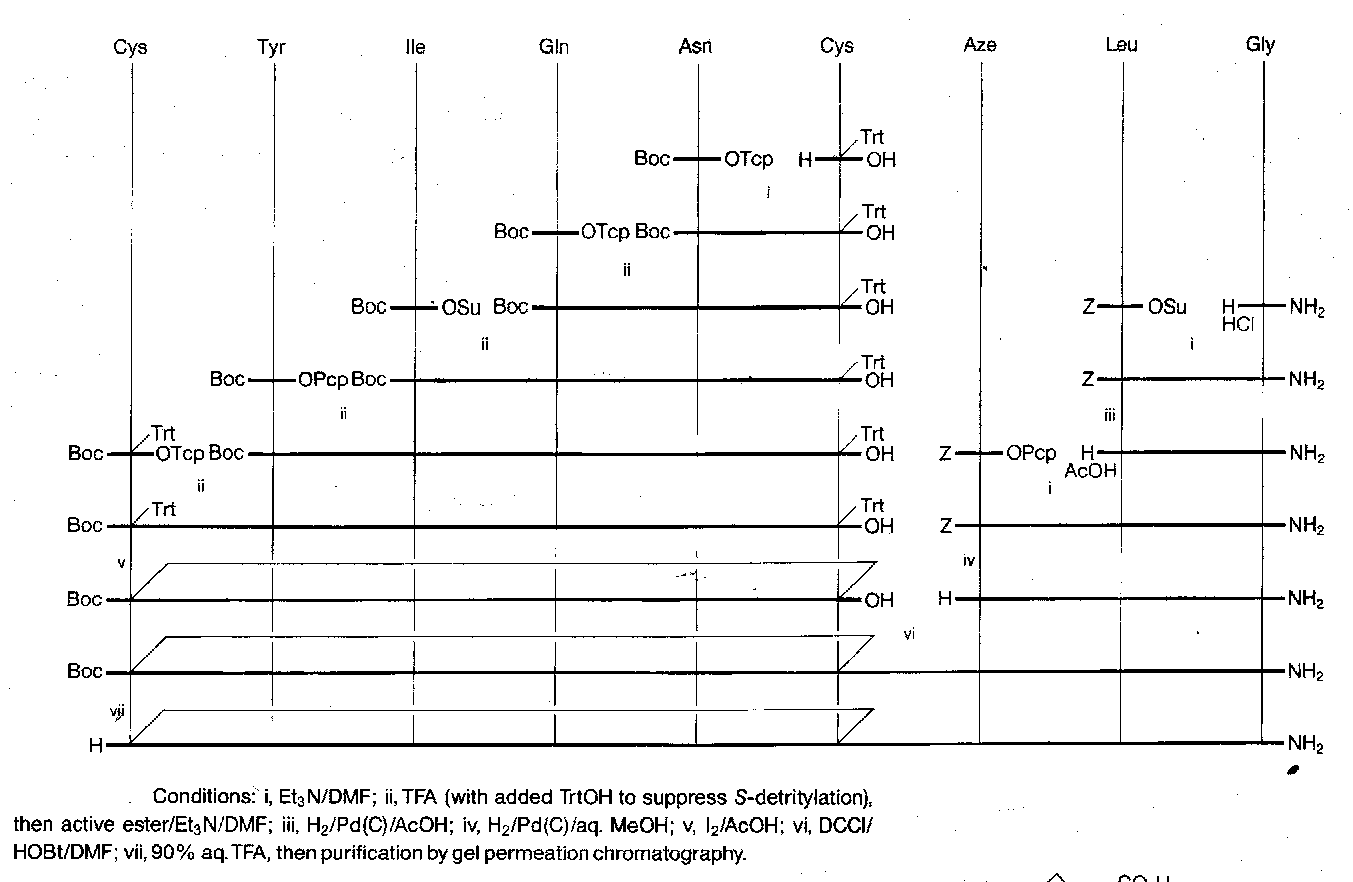
Human Insulin
This is made up of two chains:
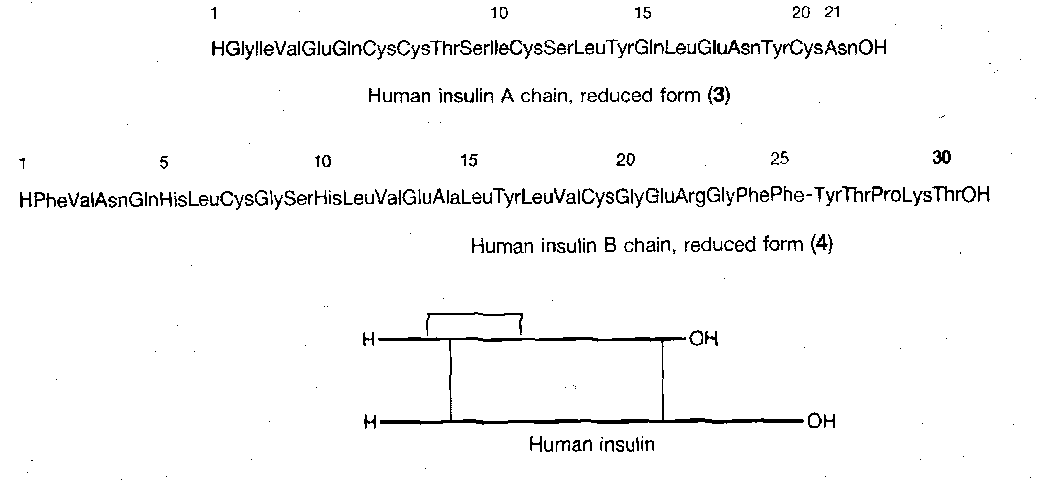
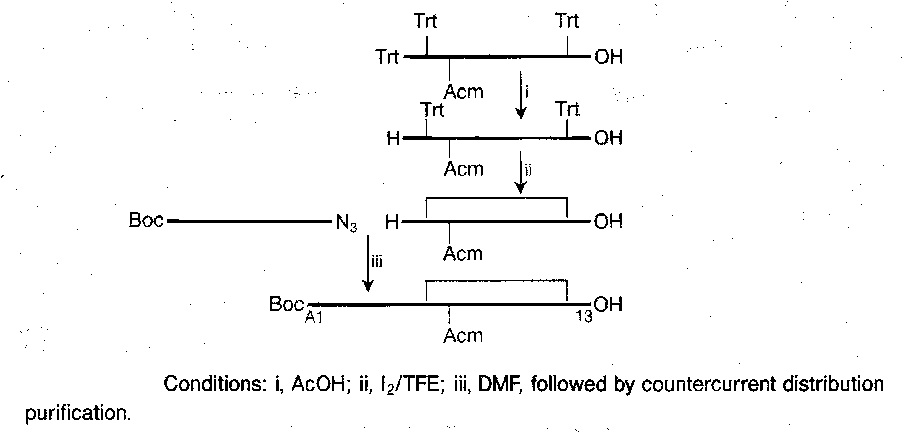
It is tricky to join the chains by the disulphide bridges in the correct places. A suitable synthesis is shown below. The key features are the selective removal of a trityl in the presence of Bpoc, the use of sulphenyl thiocarbonate method to make the unsymmetrical two-chain disulphide starting material, and the selective formation of a disulphide bridge between two Cys(Trt) residues in the presence of Cys(Acm).
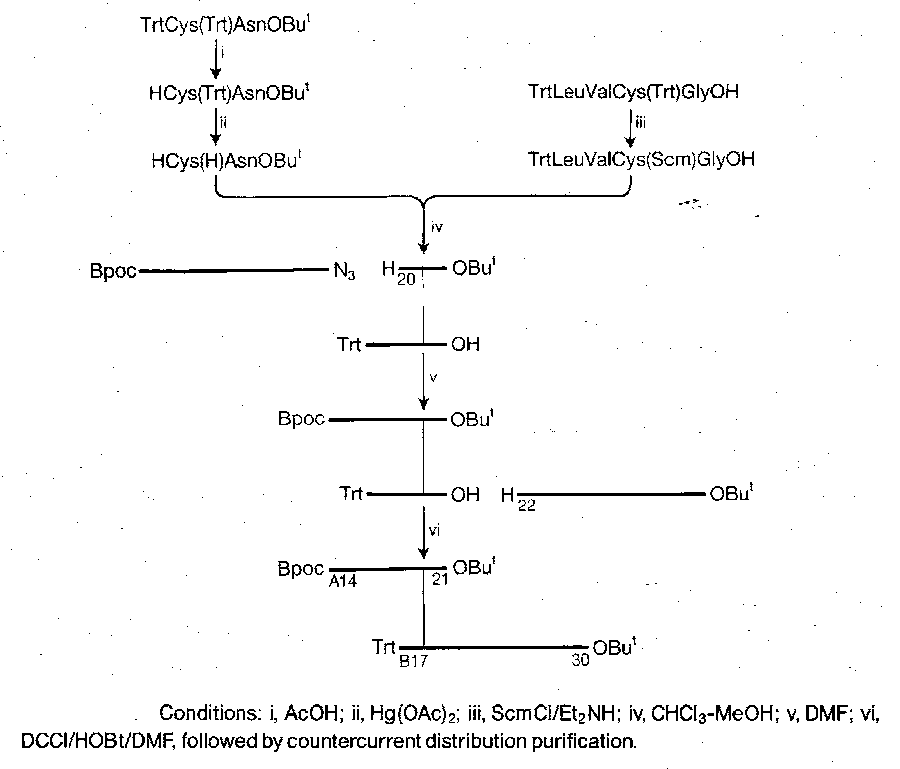
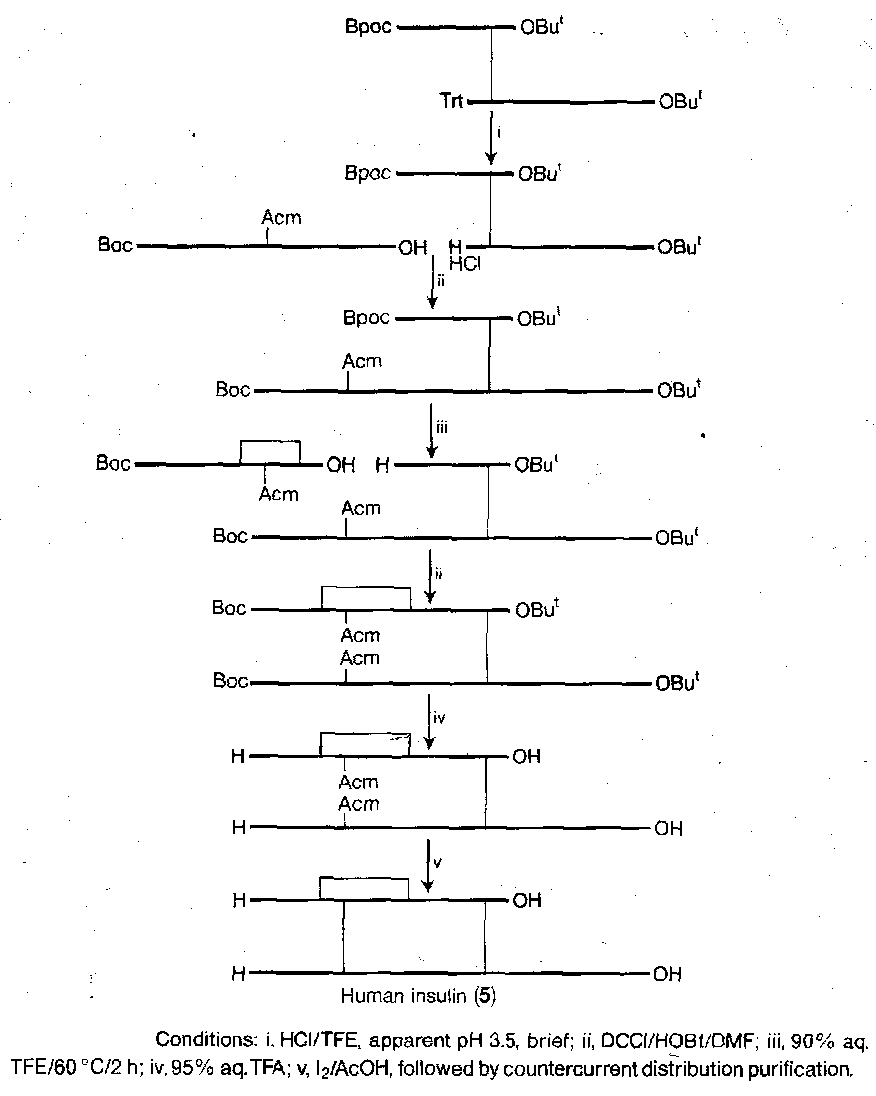
Solid Phase Peptide Synthesis
First detailed by Merrifield in 1963. The principle is outlined below:
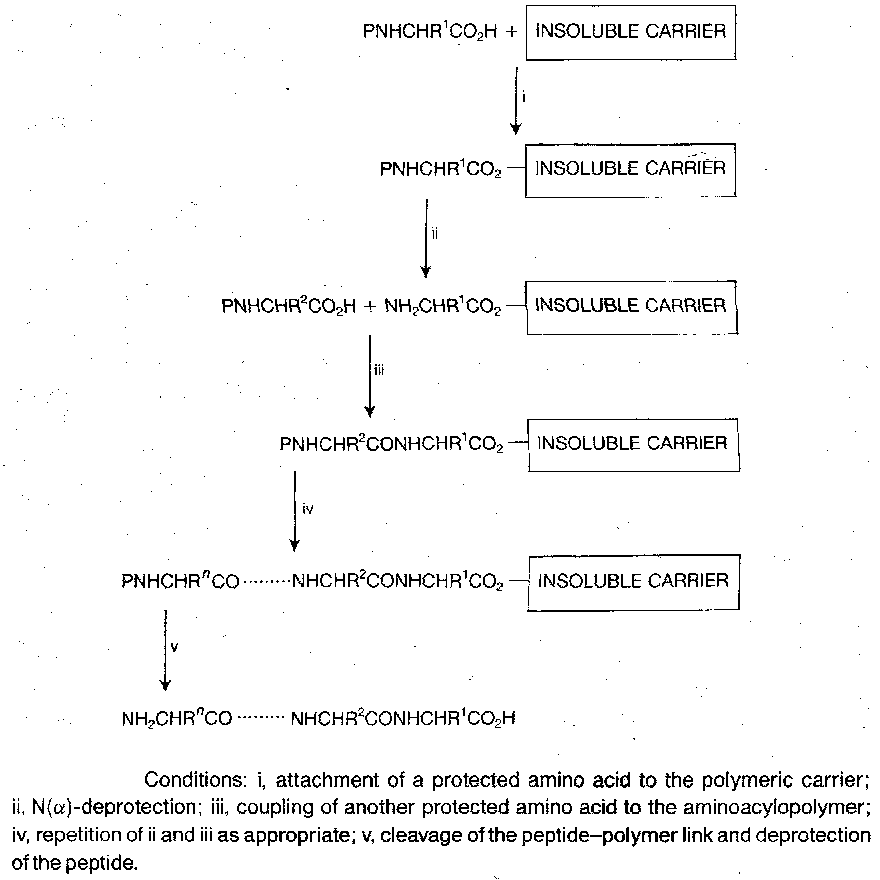
“Solid” phase is a little misleading – it takes place in swollen gel system produced by the penetration of solvent and solute molecules right into the polymeric matrix. The technique has been scaled up to industrial levels (highly automated).
The Merrifield Approach
Polystyrene cross-linked.
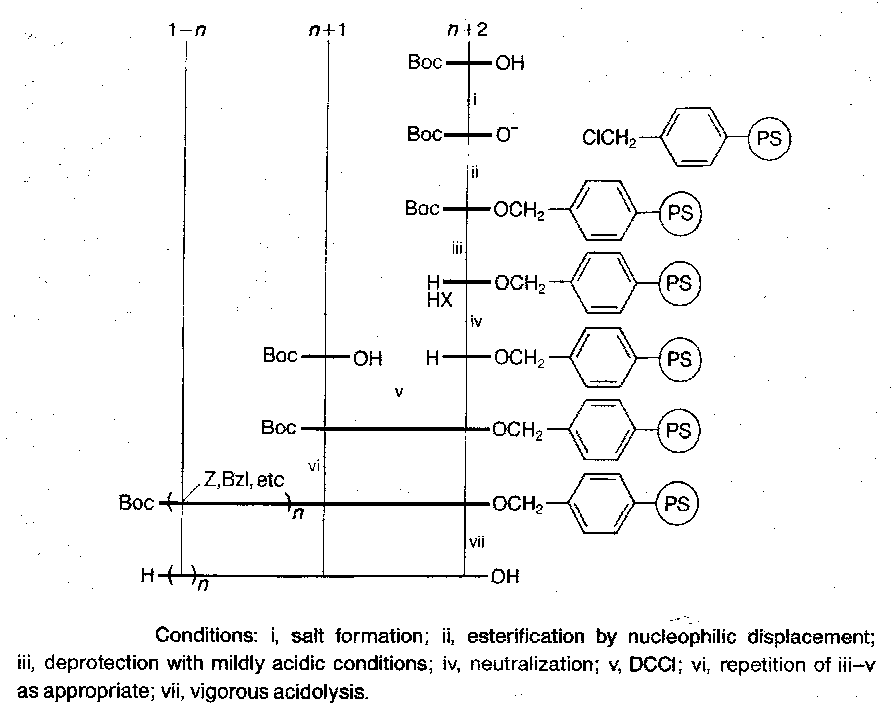
Main problems relate to:
- non-quantitative reactions.
- Incomplete orthogonality between the temporary blocking and permanent (which include the polymer link).
- Side-reactions, especially at the final complete deprotection and cleavage from the carrier.
If any N-terminal residue of the peptide-polymer conjugate remains unacylated after any coupling stage, this causes incomplete and inaccurate sequences. Hence, extensive washing is carried out between each stage.
In favourable cases, coupling can be pushed to very nearly 100% by use of excess acylating agent, or performing the stages more than once. The use of highly activated intermediates may be advantageous. Completeness can be monitored by colour tests for residual amino groups. Advanced separation and characterisation technology have been extremely helpful in dealing with the difficult purification problems.
In the standard Merrifield approach, the differentiation between the temporary α-amino protection on the one hand, and the permanent side-chain protection and peptide-resin link on the other, is dependent on the much greater acid-lability of Boc groups than benzyl ester and related groups. The difference is perfectly sufficient for syntheses involving a modest number of steps, but slight side-chain deprotection of Lys(Z) residues can occur under Boc-cleavage conditions, leading to the formation of branched peptides, and some loss of peptide from the peptide-resin conjugate takes place at each Boc deprotection, because the benzyl ester anchor is not entirely resistant to acidolysis.
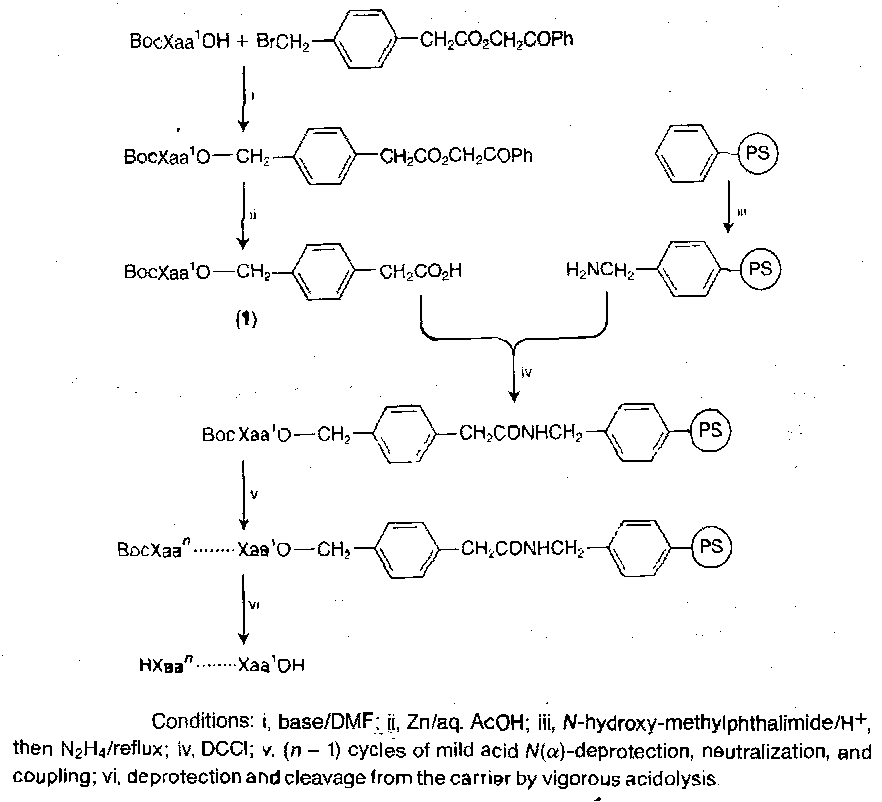
Many biologically active peptides have an amide function at the C-terminal.
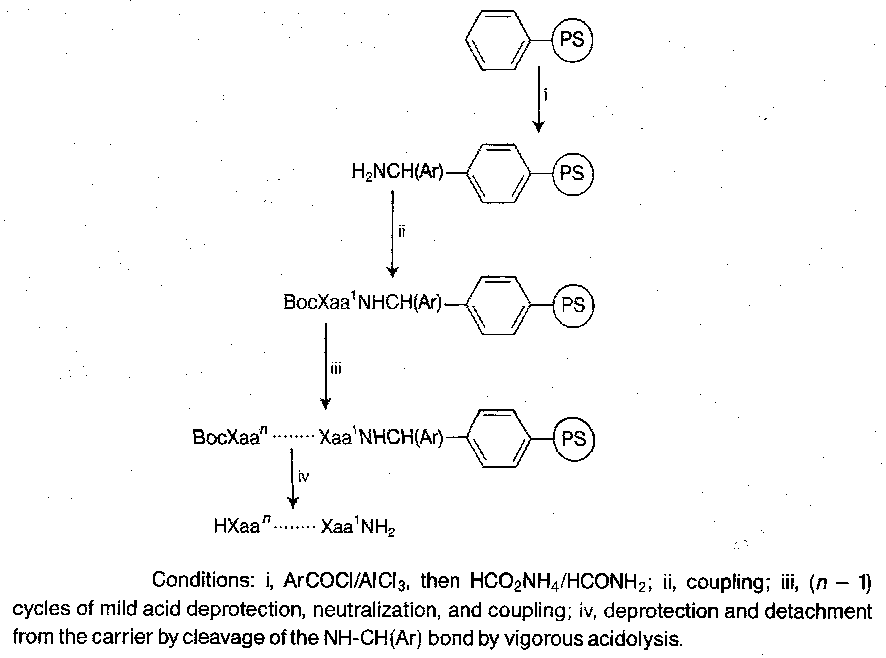
Case-Study – Bradykinin

The Sheppard Approach
In 1971 Sheppard argued that it would be desirable for the polymeric carrier to be chemically similar to the growing peptide chain, so that both carrier and peptide could be well solvated by dipolar aprotic solvents, to give an open gel system in which there was free access of reagents to reactive sites. This was in contrast to the situation with peptides attached to Merrifield cross-linked polystyrenes, where the favoured solvent for swelling the polymeric matrix (DCM) was unable to solvate the conjugated peptide chain well and thereby encouraged its aggregation, reducing the availability of reactive groups. Hence, polyamide carriers were introduced. Fmoc protecting groups were also introduced in place of Boc for greater orthogonality.
The standard Sheppard carrier is derived from a cross-linked polyacrylamide incorporating a number of sarcosine methyl ester side-chains. The side-chains are extended by treatment with ethylenediamine, followed by coupling with Fmoc-norleucine, which provides an internal reference amino acid. A “linker” group or resin “handle” is added after Fmoc cleavage, and the first protected residue of the peptide target is attached to the exposed hydroxy function on the linker. Alternate deprotection and coupling of Fmoc residues with acid-labile side-chain protection assembles the required sequence, which is then α-deprotected and detached from the carrier with simultaneous side-chain deprotection, by mild acidolysis.
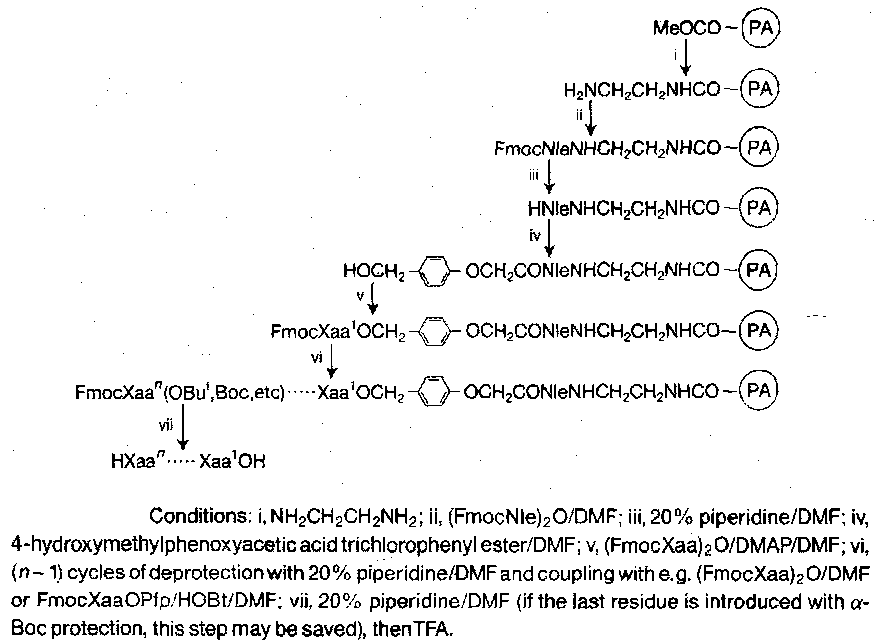
The acid-lability of the peptide-carrier link is due to the 4-alkoxybenzyl ester. Other options are available to suit. The α-Fmoc, ω-mild-acid-labile strategy has advantages over the Merrifield (α-Boc, ω-strong-acid-labile) in two ways:
- the α/ω protection is absolutely orthogonal
- the final deprotection can be carried out under much gentler conditions.
Furthermore, a single solvent, DMF, is generally used at all stages, reducing the number of washes needed, and avoiding difficulties due to swelling and contraction of the polymer with solvent.
A common and serious problem in solid phase synthesis arises from the tendency of certain sequences to self-aggregate on the resin. This reduces the accessibility of the N-terminal group to reactants in solution, which interferes with coupling, deprotection, and monitoring methods based on end-group detection. The problem usually appears after six or seven residues have been added, when it becomes necessary to use extended reaction times, repeated reaction cycles, extremely reactive acylating agents, or aggregation-disrupting media in order to continue chain assembly. It is difficult to predict when this will happen (or not).
Case Studies
Acyl Carrier Protein-(65-74)-decapeptide
Synthesised by Sheppard Method where the Merrifield failed, probably because of aggregation-related phenomena. Uses pentafluorophenyl ester activation.
|
Carrier |
As above |
|
N(α)-Protection |
Fmoc |
|
Side-chain protection |
Asp(OBut), Tyr(tBu) |
|
Attachment of the first residue |
(FmocGly)2O / DMAP / DMF |
|
N(α)-deprotection |
20% piperidine in DMF |
|
Coupling |
FmocXaaOPfp / DMF, with catalysis by HOBt in some cases |
|
Cleavage from carrier and side chain deprotection |
Fmoc removal as usual, then 95% aq. TFA. |
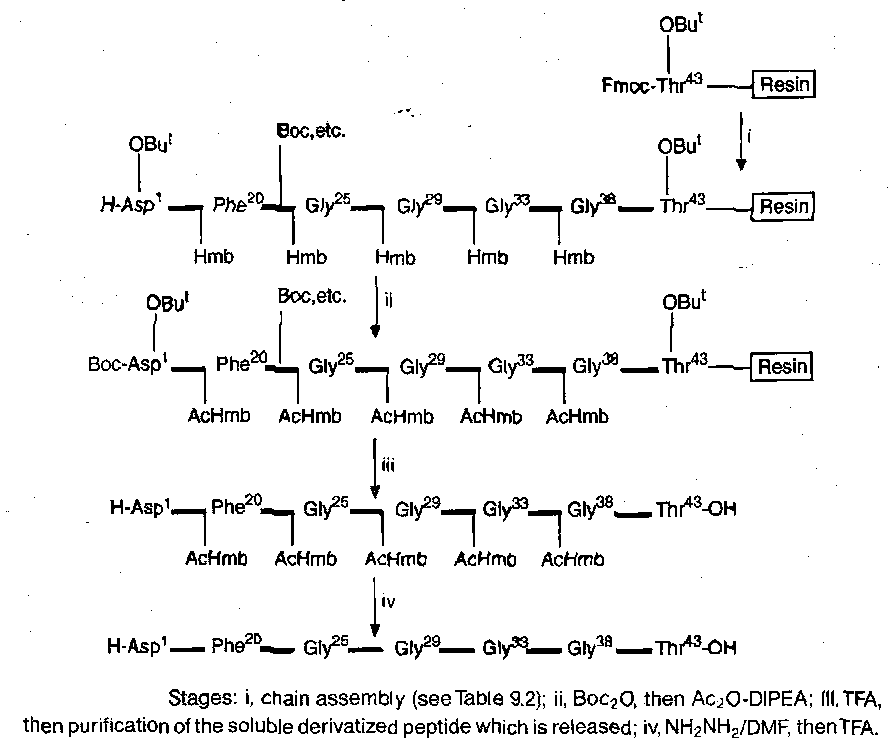
β-amyloid (1-43) peptide
|
Starting Material |
FmocThr(But)-Pepsyn KA, i.e. FmocThr(But) esterified to a 4-alkoxybenzyl alcohol, which was linked through the alkoxy group to a norleucine residue, and thence to a polydimethylacrylamide. |
|
N(α)-protection |
Fmoc |
|
Side-chain protection |
Lys(Boc), Ser(But), Thr(But), Tyr(But), Gln(Trt), His(Trt), Arg(Mtr) |
|
N(α)-deprotection |
20% piperidine in DMF |
|
Coupling |
FmocXaaOPfp / DMF in most cases, except after an Hmb-residue, when a Fmoc NCA/CH2Cl2 was used to add the next residue |
Comparison of Solid Phase & Solution Synthesis
Generally speaking, Solid Phase Synthesis is adopted for medium sizes (Sheppard generally predominating), while proteins are assembled in solution from fragments made by Solid Phase methods.
Solution Synthesis is harder to wash, as in the Solid anything not attached to the polymer chain is washed away, giving a purer product. However, for the solid, must ensure 100% protection / deprotection at each step, otherwise missing residues attach.
I am happy for them to be reproduced, but please include credit to this website if you're putting them somewhere public please!