Oxidation and Reduction in Organic Synthesis
Divided into Oxidation then Reduction. Covers the lecture notes, with more basic useful conversions thrown in (which are actually more helpful!)
Oxidation and Reduction
Introduction
In general, reducing agents are electron-rich systems and compounds – such as carbonyl groups – which are easily attacked by nucleophiles and undergo easy reductions. In contrast, oxidising agents are typically electron-deficient species, so that compounds which undergo easy electrophilic attack will also be easily oxidised.
Oxidation
Oxidation By Metal Ions
Metal oxidants can be considered mechanistically as either:
2 electron oxidants – Cr(VI), Mn(VII), Os(VIII), Tl(III), Pb(IV), or
1 electron oxidants – Fe(III), Cu(II), Ce(IV).
Oxidation of Alcohols and Related Species
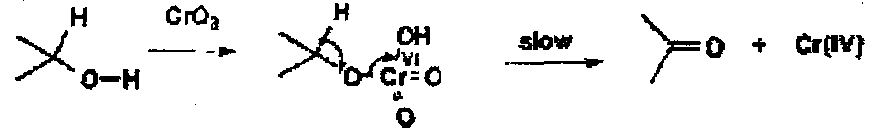
The oxidation of alcohols by Cr(VI) goes by initial fast formation of a chromate(VI) ester, followed by rate determining break down (shown by a primary kinetic isotope effect) to the ketone and Cr(IV). The details of the chemistry are complex since Cr(IV) disproportionates into Cr(V) and Cr(III) and many of these other species are 1, 2 or 3 electron oxidants.
Classic oxidation rates of axial versus equatorial alcohols:
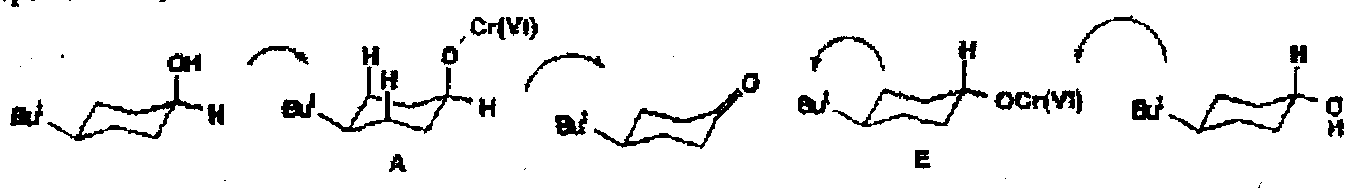
Axial alcohols are oxidised faster than equatorial alcohols since the rate determining breakdown of the axial intermediate A is accompanied by relief of 1,3-diaxial interactions not present in the breakdown of E.
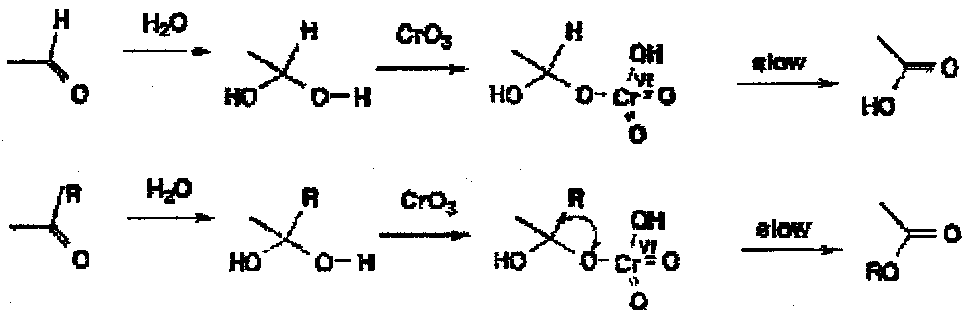
One problem is that carbonyl compounds tend to get oxidised further, usually by the same mechanism on the hydrates or hemiacetals; thus aldehydes form acids whereas ketones give esters. There are other mechanisms for further oxidation, for example by the enol, but it is clear that the absence of water would remove the above as a potential pathway for over-oxidation.
To this end a number of reagents have been devised which allow the Cr(VI) reagent to be used in solvents such as dichloromethane from which water be excluded.
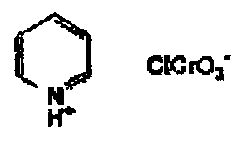
PCC (pyridinium chlorochromate) is the classic oxidant which is frequently used in the presence of a dehydrating agent. It is less effective in the presence of allylic substrates.

PDC is the less acidic form, with no chloro (pyridinum dichromate). This is good for allylic substrates.
Collins reagents are CrO3-pyridine complexes, particularly CrO3.2C5H5N. This does not attack alkenes.
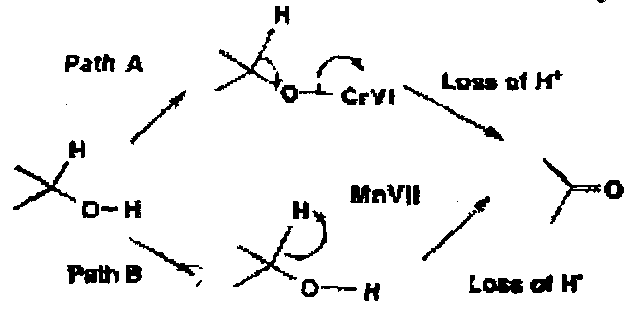
In summary, the common mechanistic path A for chromium and many other metals involves formation of a metal ester followed by rate determining breakdown with loss of a proton. An alternative path B is sometimes followed – for example in some Mn(VII) oxidations – in which the carbon-bound hydrogen is lost as a hydride ion.
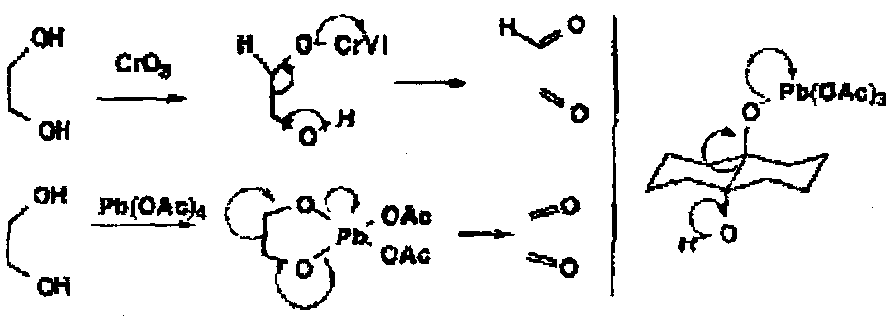
A number of alternative reactions compete with the oxidation of the alcohol, such as cleavage of the C-C bond by loss of the proton of the hydroxyl on a vicinal alcohol.
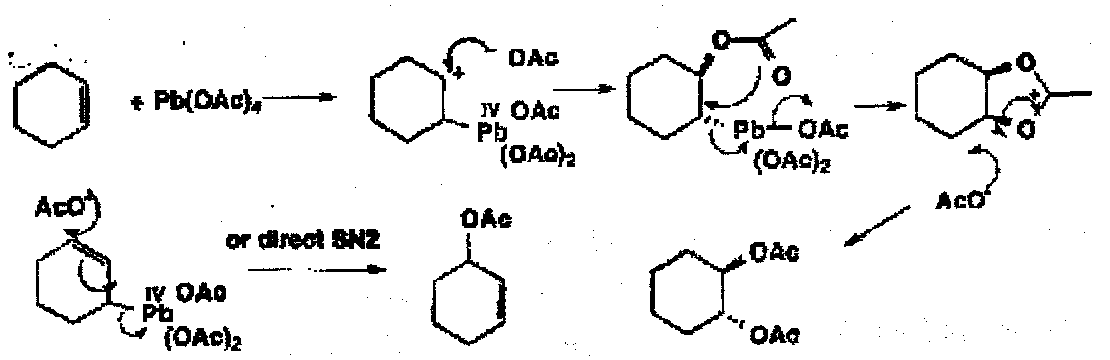
Milder two electron oxidants will also achieve this oxidation, including periodate and lead tetra-acetate which usually goes via a cyclic Pb(IV) ester.

However, even rigid trans-diaxial diols are cleaved by lead tetra-acetate via a non-cyclic mechanism.
Dess-Martin Periodinane
Hypervalent iodine. It is a mild and neutral reagent. It can be formed by adding KBrO3 to 2-iodobenzoic acid in sulphuric acid, under heat with acetone or acetic acid.
It forms the following, which oxidises alcohols to aldehydes:
TPAP
A metal oxidant very commonly used for alcohol to carbonyl transformation is Tetra-isopropyl ammonium perruthennate (TPAP) usually used catalytically in the presence of an amine oxide as the re-oxidant – no mechanistic proposal on the course of the reaction has yet been published.
Pt/O2
This can be regulated effectively, but requires heats up to 320oC. It is the reverse of hydrogenation. Alkenes are unaffected. It preferentially favours primary alcohols (1o > 2o > 3o), and stops at the aldehyde.
Manganese (IV) Oxide
Selective for allylic and benzylic hydroxyls. It works in a neutral solvent at room temperature. It proceeds to the aldehyde, unless cyanide is present.
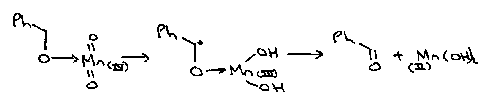
Silver Carbonate (Fetizon)
Primary or Secondary alcohol reacts to form aldehyde or ketone. It is non-destructive.
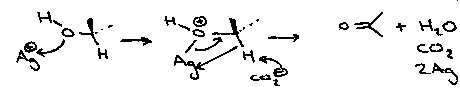
It is concerted and reversible. Adding NaOCl means that secondary alcohols are oxidised before primary. Polar solvents generally will inhibit the reaction.
Oppenhauer
Al(OiPr)3 in acetone (or any H acceptor).
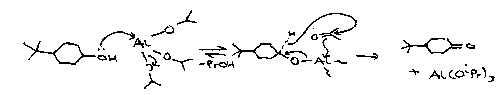
The reverse is Meerwein-Ponndorf-Varley mechanism, a REDUCTION.
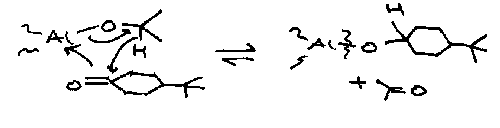
Carboxylic Acids
As mentioned, it can sometimes be difficult to stop reactions proceeding past the aldehyde stage and straight to the carboxylic acid. Most of the reagents above are designed to stop at the aldehyde stage. The major ones for fully oxidising an alcohol are shown below.
RuO4 is a very powerful oxidising agent and works at room temperature, in CCl4. KMnO4 is similar.
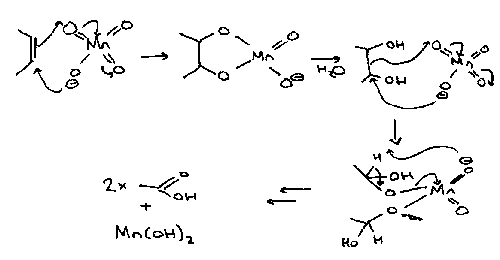
Jones’ Reagent –
CrO3 in sulphuric acid and acetone. Mechanism as given for general alcohol oxidation at the start of this section.
Silver Oxides –
AgO is potent at acting on aldehydes. Ag2O is weaker, and selectively oxidises CHO to COOH. It does not react with α,β unsaturated compounds.
Amines
These are easier to oxidise than alcohols: the opening step is electrophilic substitution of the N-H by the metal to give an intermediate which can fragment in a number of ways to give a range of products, e.g. by loss of a proton to give an imine or C-N bond cleavage.
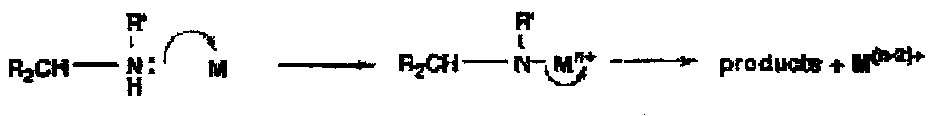
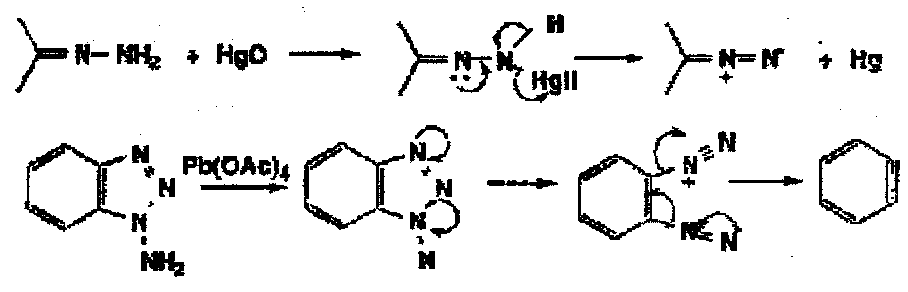
Thus these oxidations tend to be useful if a pathway is particularly easy and the oxidising agent relatively mild: for example, hydrazones are oxidised by Hg(II) to diazoalkanes and the aminotriazole is oxidised to benzyne by lead tetra-acetate at -78oC.
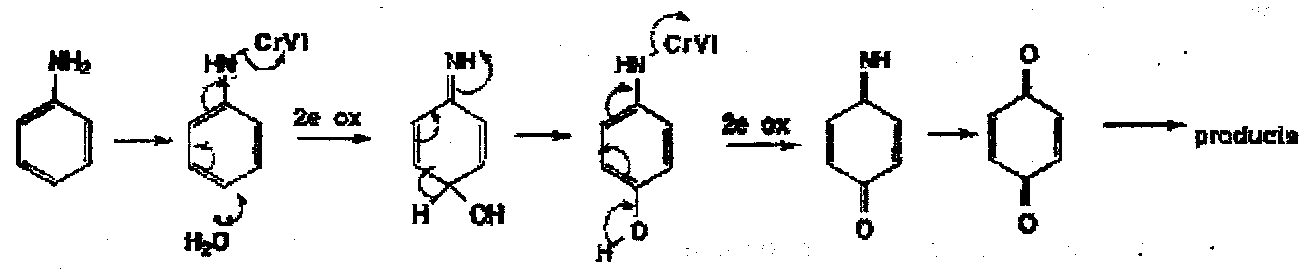
These are easy oxidation of both aromatic amines and phenols by 2 electron pathways: among other mechanisms the following is probably one of several paths in the oxidation of aniline by Cr(VI):
Aromatic amines are also easily oxidised by 1 electron oxidants such as hexacyanoferrate(III), a way of keeping Fe(III) in solution without it precipitating as rust. An initial one electron oxidation gives a radical cation, which loses a proton to give a radical. A second one electron oxidation which loses a second proton to give the p-benzoquinone.

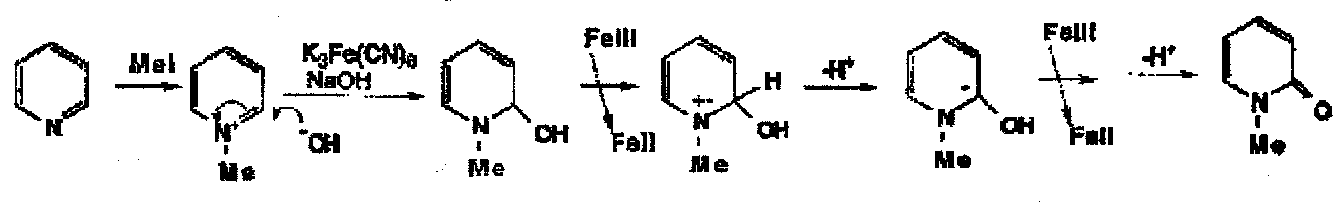
A second example is the oxidation of N-methylpyridinium cation in base to N-methylpyridone, again involving sequential loss of an electron and a proton twice:
Phenols, usually as the phenoxide ion, also undergo easy oxidation by Fe(III). A famous case is the oxidation of p-cresol to a radical which dimerises. A subsequent intramolecular Michael reaction gives Pummerer’s ketone.

The mechanism of the formal dimerisation may be rather different than that indicated.
Another example is Fenton’s Reagent, Fe(II) and H2O2, i.e. Fe(III) and –OH, which oxidises α-hydroxy acids to aldehydes via an oxygen-stabilised radical.
In summary, the oxidation of a neutral compound by a one electron oxidant initially forms a radical cation which loses a proton to give a radical. One electron oxidation of an anion gives a radical directly.
Oxidation of Alkenes
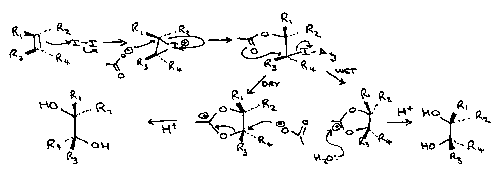
Prevost
Varies under dry or wet conditions, as water can act as a nucleophile.
Dry conditions leads to the ANTI product, while Wet conditions favour SYN. This is shown by the mechanisms:
Alkene Cleavage
Can be affected by OsO4 (diol), RuO4 (COOH), MnO4- (COOH), CrO3.
Metal Ion Oxidations
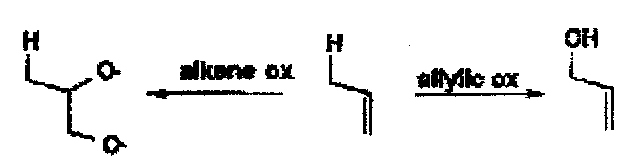
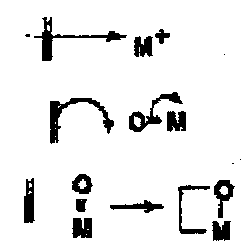
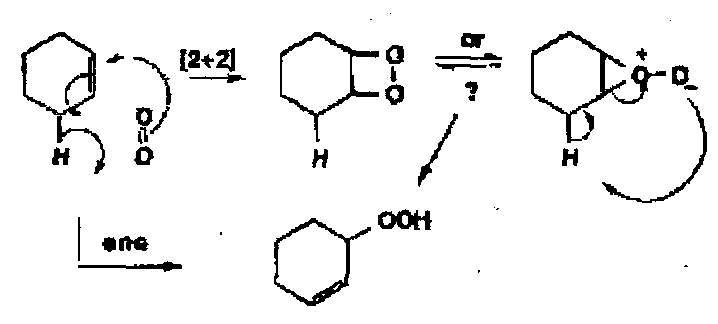
Oxidation of alkenes can result either in allylic oxidation or oxidation of the C=C. The mechanism can be either a 1 or 2 electron process and the initial interactions between the alkene and the metal are:
- Initial electrophilic attack on the alkene by the metal, followed by many different paths, including possible concerted ene-type reactions.
- Occasionally direct attack on the oxygen.
- Reaction with the metal peroxo species in a formal [2+2] cycloaddition to give a metallo-oxocyclobutane as a first formed intermediate.
The detailed mechanisms of these pathways has not been clearly demonstrated.
Transition Metal Catalysed Epoxidation of Alkenes
Two of the outstanding advances in asymmetric synthesis have been due to Sharpless and are both oxidation of alkenes by peroxometal species by equivalents of Os=O for cis-hydroxylation and Ti=O for epoxidation. The principal of this is:
- to look for ligand acceleration of a metal-mediated process, and
- look for the right chiral ligands.
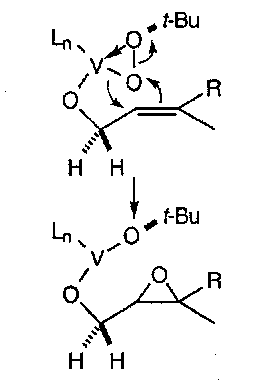
The use of transition metals in conjunction with organic peroxides has yielded a very useful set of oxidising agents which show amazing selectivity for the oxidation of alkenes with an allylic hydroxyl group.
By adding a chiral ligand, a chiral oxidant is formed:

Titanium tetraisopropoxide is the most effective transition metal complex and diethyltartrate (DET) is the best chiral ligand. The reaction that ensues between an allylic alcohol and the chiral complex gives a single enantiomer. It works on most alkenes, although 1,2-cis-disubstituted alkenes do not always give high enantioselectivity. It is also good because it only uses catalytic quantities of the transition metal.
The reaction proceeds via an active complex which is almost certainly a dimer in which each Ti atom behaves independently.
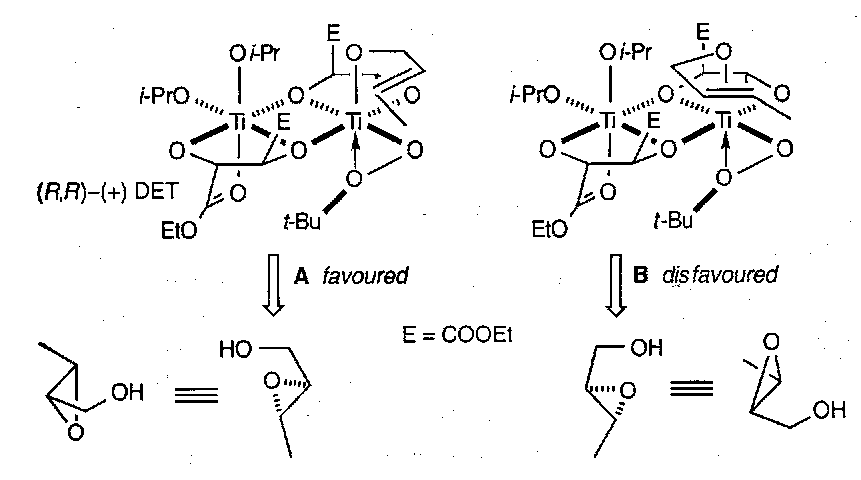
The allylic alcohol on the right-hand side of the dimer has an alkene which adopts a conformation whereby it can lie above the active oxygen of the peroxide. A is favoured over B due to sterics.
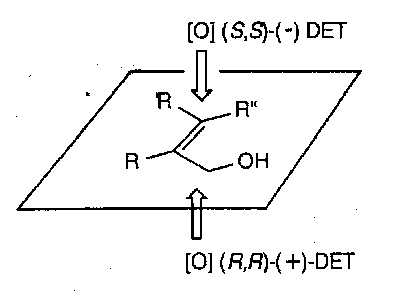
The Sharpless epoxidation is so reliable that a general rule has been developed to help chemists determine which enantiomer of DET will produce which epoxide. Drawing the allylic alcohol in the orientation illustrated below, and (-)-DET epoxidises from the top face and (+)-DET from the lower face.
Osmium Tetroxide Oxidations
The basic mechanism probably involves an initial formal [2+2] cycloaddition to give a metallo-osmacycle which then undergoes a ring expansion to form a cyclic osmium(VIII) ester. Subsequent hydrolysis gives the cis-diol and Os(VI). Osmium is expensive and poisonous and so tertiary amine oxides such as N-methylmorpholine-N-oxide or iron(III) are used as oxidants for re-converting Os(VI) to Os(VIII).

Such changes can complicate the mechanism: for example does the Os(VI) ester get hydrolysed before it is re-oxidised, or does it get oxidised first? The details are still being sorted out. The key observation was: some amines accelerate the reaction (by complexing to Os), though some amines slow it down. If you have an amine which is chiral and accelerates the reaction, this will cause asymmetric induction and be a good candidate for a catalytic asymmetric hydroxylation.
If the amine is achiral, then the transition states are enantiomeric and so, since they have the same energy content, equal amounts are formed so you get a racemic mixture. If the base is chiral, the transition states are diastereomeric and thus different amounts of two enantiomers are formed.

The original ligand base was a quinine derivative – there are now some more designed bases and the scope of the oxidation is now almost general.
Titanium-Catalysed Epoxidations
Only for allylic alcohols since complexation to Ti reagents is necessary.
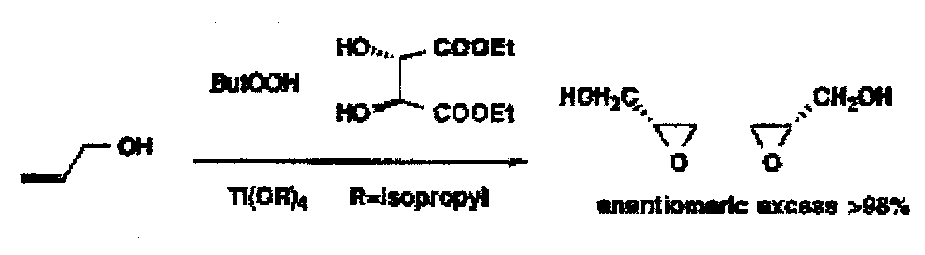
Reagents: titanium isopropoxide as catalyst, diethyl tartrate as the chiral ligand, tert-butyl hydroperoxide as the oxidant.

- An equimolar complex of titanium isopropoxide and the tartrate diester C is the catalytically active template; it is much more active than titanium propoxide itself or of titanium tartrates of other than 1:1 stoichiometry, and thus exhibits ligand accelerated catalysis.
- The rate is first order in substrate, oxidant and inverse second order in inhibitor alcohol under pseudo-first order conditions in catalyst – this is characteristic of a system in which the allylic alcohol and alkyl hydroperoxide bind to the same metal centre.
- The rate of epoxidation is slowed by alkenes with electron-withdrawing groups, indicating the allylic alcohol moiety is nucleophilic.
- Increased bulk at several positions in the epoxidation system results in increased epoxidation rates together with better kinetic resolution and asymmetric induction.
More reactive metal peroxo compounds, e.g. V(V)=O and Mo(VI)=O will epoxidise most alkenes.
Alkenes with metals go to initially π or σ complexes. With fairly weak 2e oxidants such as Tl(III), Hg(II) or Pb(IV), fairly selective oxidations take place.
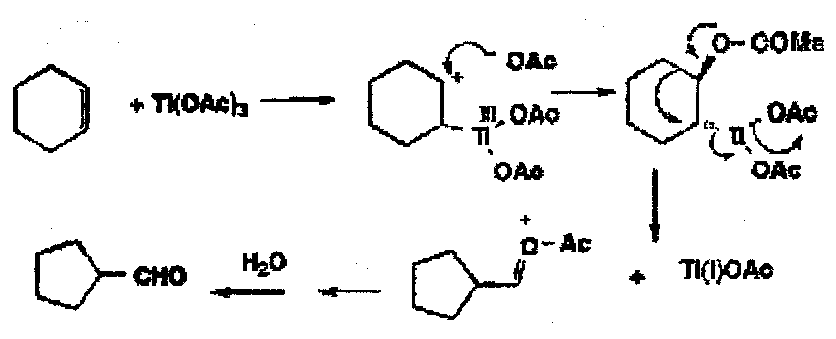
Tl(III) oxidises cyclohexene to cyclopentene aldehyde by initial trans addition with a subsequent oxidative ring contraction with loss of Tl(I). In the case Pb(IV) acetate, a number of different fates of the first formed carbocation give several different products, including allylic acetoxylation and the formation of a trans-cyclohexane diol.
Chromium and Manganese in their higher oxidation states are stronger oxidising agents and give other products. Cr(VI) gives an ene product which then undergoes a [2,3]-sigmatropic shift to give a species which can disproportionate or hydrolyse. With Cr there is then further oxidation. However, with Selenium dioxide, which goes via similar pathway, the allylic alcohol and enone products are usually formed in good yields. The Se(IV) oxidation of ketones to give 1,2-diketones goes via an analogous pathway.

In summary, in order to rationalise most of the metal oxidations of alkenes, the best plan is to draw the initial σ complex and then think about the likely fates.
Oxidation by Oxygen and Related Species
Triplet (ground state) oxygen: autoxidation
Oxygen is a diradical and itself is a good radical trap.
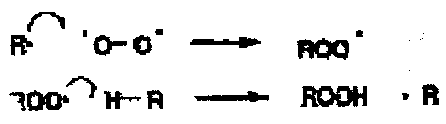
The propagating steps for autoxidation are coupling between the organic radical and triplet oxygen to give a peroxyalkyl radical which abstracts a hydrogen from the alkane to regenerate the alkyl radical. The second step, in which a strong O-H bond is formed from a C-H bond will be more exothermic when the R-H bond is weak, i.e. when the carbon radical is more stable.
In the autoxidation of cyclohexene, a stable allylic radical is an intermediate.

Allylic autoxidation is frequently a nuisance, so there is much effort in understanding methods of inhibition.
Some common solvents such as ether and chloroform, both of which form stable radicals by removal of a hydrogen, give hazardous autoxidation products.
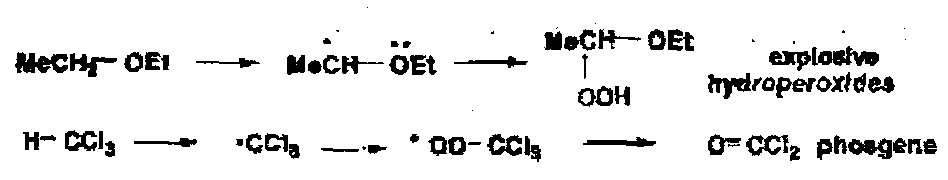
It is clearly necessary to inhibit this and ethanol is added; EtOH is an excellent radical trap due to the weak C-H bonds.
Singlet Oxygen
Formed by a number of methods, is commonly formed by photolysing triplet oxygen in the presence of a dye such as Rose Bengal which allows intersystem crossing. Singlet oxygen is a reactive intermediate which does many pericyclic reactions.
For example, with cyclopentadiene at -78oC, a Diels-Alder reaction gives a strained endoperoxide which, as it warms to room temperature, fragments the weak O-O bond.
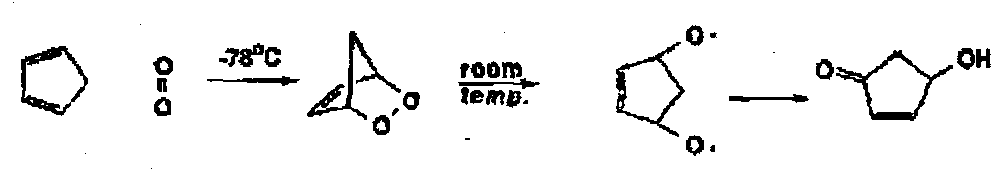
Singlet oxygen will also do a [2+2] cycloaddition to give either the dioxetane or an oxide perepoxide, both of which may occur and may be equilibrating – they both will give the cyclohexenyl hydroperoxide as product. In competition with these reactions, a concerted ene reaction can give the same product.
The different pathways may depend on substrate and conditions so that the details of the mechanism are complex.
Organic peroxides and peracids are common intermediates, in particular for reactions in which there is a migration to e-deficient oxygen. Examples of this are:
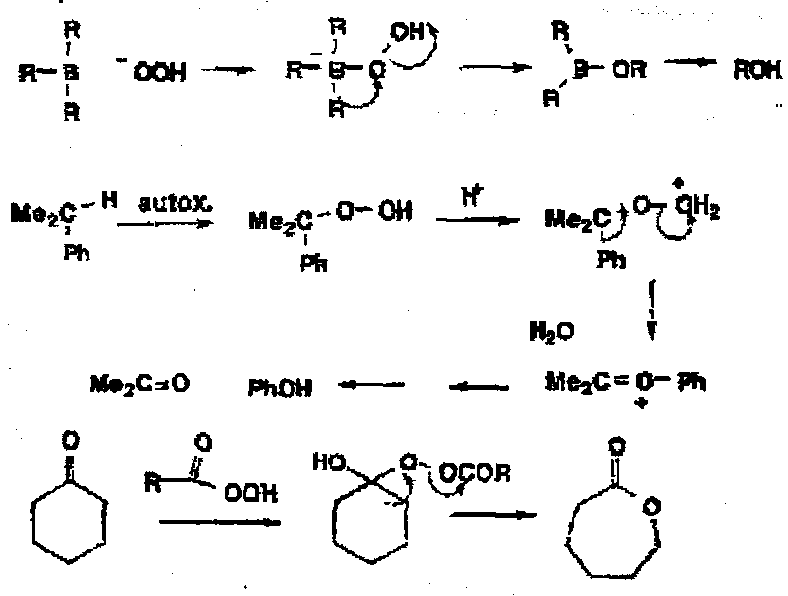
- The oxidation of alkyl boranes by alkaline hydrogen peroxide to give an anionic intermediate which fragments with loss of hydroxide ion.
- The industrial preparation of phenol which involves autoxidation of cumene to the hydroperoxide which with acids loses water giving the e-deficient oxygen species with migration of the aromatic ring to give acetone and phenol.
- Baeyer-Villiger oxidation of a ketone with a peracid to give an adduct which then fragments with loss of carboxylate and migration to the e-deficient O, in the case of cyclohexanone giving a ring expansion to a seven-membered lactone.
Peracids oxidise C=C to epoxides in a concerted reaction.

The nitrogen analogue, formed in situ from hydrogen peroxide and a nitrile, is a stronger epoxidant. Dimethyl dioxirane and N-sulphonylazairidines are some new-ish reagents which also epoxidise.
For electron poor C=C, such as cyclohexenone, the usual reagent is alkaline hydrogen peroxide in which the opening step is nucleophilic addition across the enone followed by intramolecular cyclisation to form the epoxide.
Ozone provides a classic case of 1,3-dipolar cycloaddition reaction.
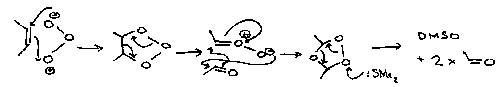
An initial cycloaddition (A) is followed by a cycloelimination to give another 1,3-dipole, a carbonyl oxide, as a real trappable intermediate. A final cycloaddition (C) gives the isolated ozonide. Reductive work-up with dimethylsulphide gives the two carbonyl compounds.
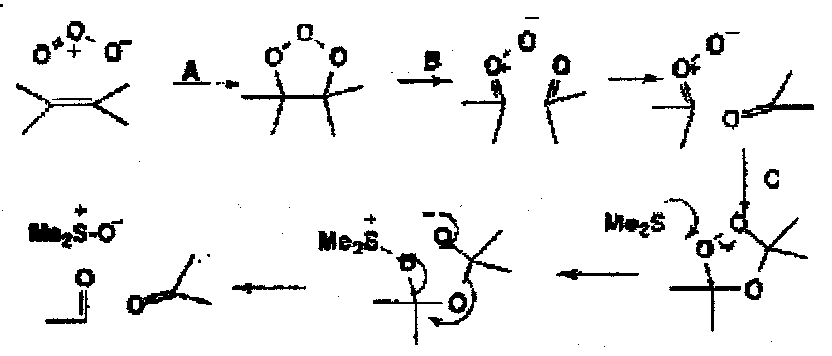
The reaction is excellent and highly selective and is very useful in synthesis. Ozone is also a hydride acceptor from some metals.
Sulphur-based Oxidants
There are a number of sulphur-based oxidants which are used for the oxidation of alcohols to carbonyl compounds which all rely on the formation of a oxosulphonium intermediate which in the presence of base breaks down, either concertedly or by proton removal and a sulphoxide-type elimination. At present, the most common procedure is the Swern oxidation.
Swern
This activates the DMSO, but requires a moderating solvent to prevent explosion. This then reacts in a similar way to Kornblum:
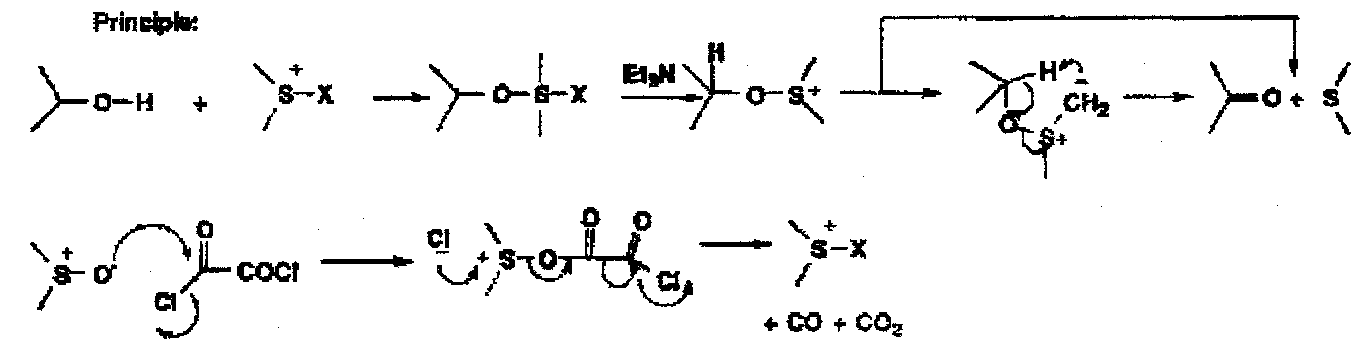
In this, a sulphur-based intermediate is generated from the alcohol, oxalyl chloride and dimethyl sulphoxide at -78oC, and then a base, triethylamine, is added to fragment the intermediate to the products.
Moffatt
DMSO not acting as a nucleophile, e.g. DMSO + DCC + H+.

Kornblum
DMSO as a nucleophile. Particularly used for Carbon-Halogen bond cleavage.
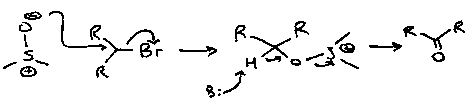
Sulphur Oxidation
R2S will react with peracids:
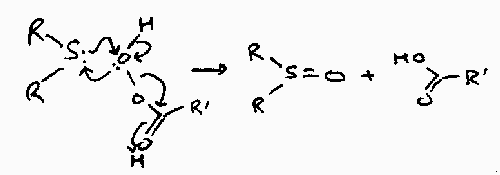
This can then be repeated to form the sulphone, R2SO2, but this requires more vigorous conditions because sulphoxide is a weaker nucleophile.
Selenium compounds can be oxidised in the same way.
Other Oxidation Reactions
Etard Reaction
CrO2Cl2 with Zn workup takes methylated aryls to aryl aldehydes.
This works by using the CrO2Cl2 with CS2/CCl4 to form chloroketones, and the addition of Zn then removes the Cl to give the ketone.
Barbier-Wieland
Removes a single carbon atom. It proceeds by first esterification, then addition of PhMgBr. This is removed using acid, and finally oxidative cleavage occurs.
Reduction
Typically, compounds easily attacked by nucleophiles will be easily reduced, since both nucleophiles and reducing agents are electron-rich.
Reduction by Complex Hydrides
Complex hydride reducing agents can be classified by their initial interactions with the substrate of the reduction as either (a) nucleophilic, (b) electrophilic, (c) radical.
Nucleophilic Reducing Agents
Chemoselective of hydride attack on carbonyls. The following is the decreasing order of reactivity of carbonyl functions with nucleophiles, together with the usual reactivity of the hydride reducing agents:
|
Carbonyl |
Acid Chloride |
Aldehyde |
Ketone |
Ester |
Amide |
|
LiAlH4 |
✓ |
✓ |
✓ |
✓ |
✓ |
|
LiBH4 |
✓ |
✓ |
✓ |
✓ |
✗ |
|
NaBH4 |
✓ |
✓ |
✓ |
✗ |
✗ |
|
LiAlH(tOBu) |
✓ |
slow |
✗ |
✗ |
✗ |
The differences need explaining as well as remembering. Remember the Lewis Acid and the solvent. This list is by no means comprehensive either – some esters are easily reduced by sodium borohydride, usually those with an α-oxygen substituent. For the reduction of acyl chloride to aldehyde, you can use a deactivated nucleophilic reagent which reacts with the carbonyl of the acid chloride but will not react with the aldehyde – probably the best is (Ph3P)2CuBH4.
Mechanism of Reduction of Esters by LiAlH4
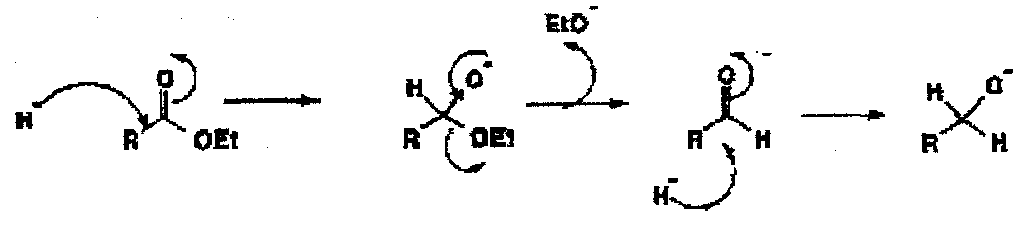

This can repeat up to 4 times. However, proceeding from carboxylic acids requires 3 H’s.
A mixture of products is often obtained when attacking 1,4-unsaturated systems, since it can donate to the alkene or carbonyl.
Since aldehydes are more susceptible to attack by nucleophiles than esters, no chance of getting an aldehyde by this process unless you can persuade the tetrahedral intermediate not to break down.
Better off using an acid chloride, in which the carbonyl group is more reactive than the aldehyde, with a more selective reducing agent such as a toned-down big bulky and chemoselective LiAlH(tOBu)3 or similar.
Mechanism of Reduction of Amides by LiAlH4

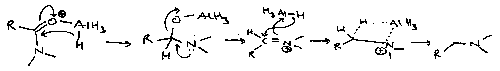
Note the leaving group is not the amine. This a major synthesis of 2o and 3o amines.

If you are able to stop at the tetrahedral adduct and only allow that to break down later, you would get an aldehyde synthesis, so you can make the break down more difficult by using either aziridine, which would introduce angle strain in the fragmentations or just stabilise with an e-withdrawing methoxy group which might be helped by chelation.
Sodium Borohydride
NaBH4 is a very versatile reagent, being very selective and less powerful, and soluble in many solvents. It is good for reducing enamines, imines et al. Also works on amides via iminium salts, acts on conjugated systems via 1,2 reduction, and reacts well with ketones.
Substituting the Metal –
This affects the reactivity and selectivity.
LiBH4 is a stronger reducing agent, and attacks epoxides, esters, ketones. It is useful in the selective reduction of esters.
Zn(BH4)2 selectively reduce α,β-unsaturated ketones, and can also reduce epoxides at the more substituted position. It also removes nitriles.
Ni, Co and Cu all favour 1,4 reductions.
Luche’s reagent is NaBH4 + CeCl3.6H2O. It selects only 1,2 reduction of conjugated ketones.
Superhydride
Bet3 + LiH forms LiBHEt3. This is the most powerful reducing agent, and reacts via SN2 mechanisms, reducing R-X.
Modifying the BH4- by making the more bulky, and sometimes incredibly bulky, R3BH- using keeps much the same chemoselectivity but has a considerable effect on the stereoselectivity.
In summary, unhindered cyclohexanones are usually attacked by hydride from an axial route giving the most stable equatorial alcohol. Hindered ketones are usually attacked by the path of minimum steric hindrance to approach which in the case of chair forms avoids bad 1,3-diaxial interactions in the transition state approach. There are marked differences in the stereoselectivity of chelating borohydrides – classically zinc borohydride – and non-chelating. It is also important to be aware of when thermodynamic or kinetic control is in effect.
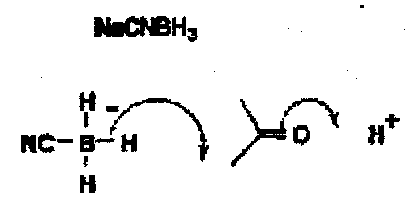
Sodium cyanoborohydride is a less good hydride donor than sodium borohydride because of the e-withdrawing nitrile coordinated to boron, and it will not reduce aldehydes and ketones at neutral pH, but will do at about pH 4 by general acid catalysis.
It is worth remembering this, since it is the classical hydride reducing agent for the reductive amination of ketones. For example, if a mixture of PhCHO and MeNH2 is treated with sodium borohydride, both the benzaldehyde and the amine are reduced, whereas with sodium cyanoborohydride at pH7 the aldehyde is not reduced whereas the iminium salt is giving a clean and unambiguous reductive amination.

Sodium triacetoxyborohydride is another reagent of choice for reductive aminations.
Electrophilic Hydride Reducing Agents
These are reagents which depend probably more importantly on the initial electrophilic interactions of the reductant with the substrate which is followed by hydride transfer. The classic reagents are borane and diisobutylaluminium hydride (DIBALH) R2AlH where the Lewis Acid character of the reagents is dominant. Thus, more nucleophilic carbonyl groups are reduced more easily by electrophilic hydrides.
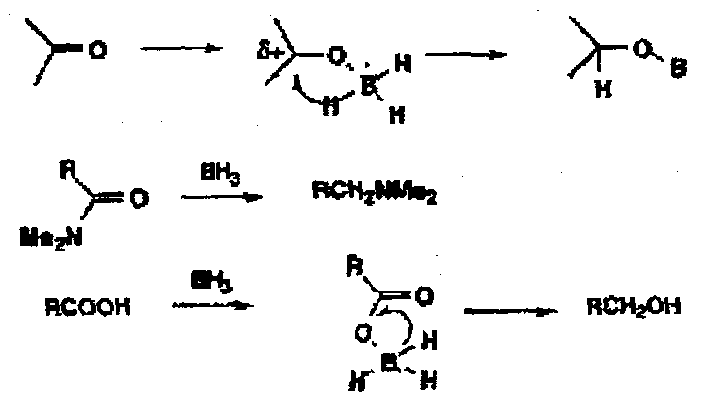
Amides are reduced to amines by boranes, but esters are usually unaffected.
Carboxylic acids are reduced under very mild conditions to alcohols, probably via an acyl borane intermediate.
AlH3
This is a strong electrophile, and selectively reduces conjugated ketones as 1,2. This is because it is a Lewis Acid and complexes to the CO (but is not large enough to reach the alkene).
DIBALH
This is diisobutyl aluminium hydride, HAl[CH2(CH3)2]2
It is highly stereoselective due to its bulk and complexing.
At low temperatures such as -78oC it reduces esters to RCHO or RCO only, while at high temperatures it reduces to the alcohol. This is because at low temperatures a stable tetrahedral intermediate forms.
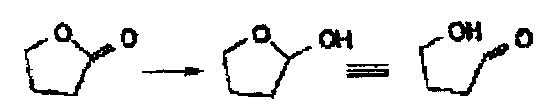
It is the reagent of choice for the reduction of esters to aldehydes (or lactones to lactols).
Tributyl Tin Hydride
This is the most common radical hydride reducing agent, but that may change for environmental/health grounds.
Bu3SnH is highly selective for halogen and can be used for the removal of halogens: this chemistry depends on the fact that M-H bonds get weaker as you go down Group 14, and the M-Hal bonds get stronger: thus you trade a relatively weak Sn-H bond for a relative strong C-H bond and also a relatively weak C-Hal bond for a relatively strong Sn-Hal bond.

The chain propagation steps are shown; it may be necessary to add radical initiators.
The Sn-S bond is also very strong and this has been used to develop the deoxygenation of secondary alcohols by the Barton Deoxygenation of xanthates.
Reduction by Dissolving Metals
These proceed via addition of an electron, then H+, then another electron, followed by another H+.
The key points in this chemistry are:
- These are almost all 1 electron stepwise reductions, and proceed from neutral substrates via radical anions as intermediates.
- Watch the solvents as to their acidity / protic – you will not reduce a function group if the solvent is reduced by the metal much more easily.
- The availability of protons very frequently determines the course of reactions.
Reduction of R-Hal

Carbonyl Group Reductions
Ketones:
An example is the Clemmenson Reaction:
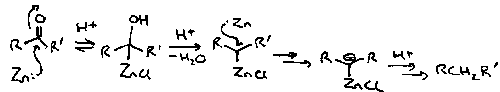
It is easier to reduce a C=O than a C=C since the resulting radical anion is more stable. Thus reduction of carbonyls can take place with a metal such as sodium in alcohol as a solvent; reduction of C=C cannot usually proceed in alcohol since the solvent is too easily reduced.

The conditions under which ketones are reduced by metals are crucial as to the products formed. Thus reduction with a metal (Na) in ethanol gives the corresponding alcohol whereas in the absence of protons, the dimerised product, pinacol, is obtained on acid work-up.
In cyclohexanones, the equatorial alcohol is always formed by axial protonation of the anionic intermediate.
Esters: again the outcome of a metal reduction of an ester depends on whether there are readily available protons. In alcohol, the primary alcohol is formed, whereas in xylene (no protons) the acyloin reduction takes place.
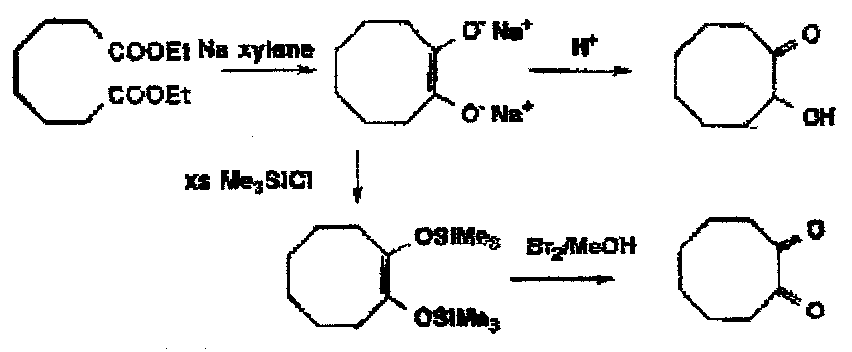
The Acyloin Condensation – sodium + xylene: the classic method for the formation of medium and large rings from acyclic materials. Mechanistically imaginative but the key feature is to watch proton availability; the end product is a di-sodium salt.
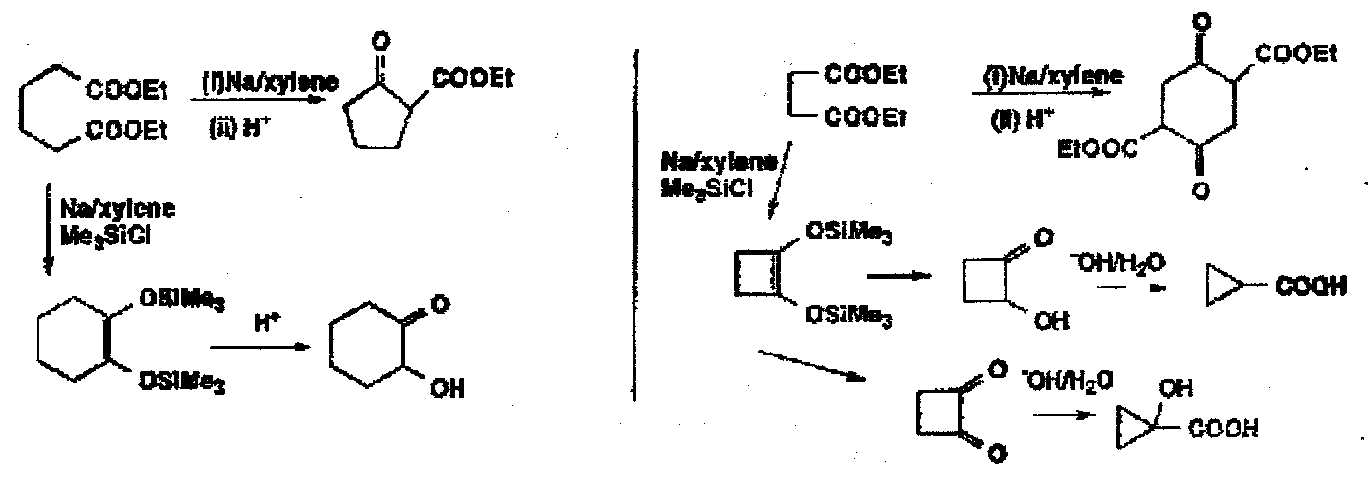
Originally bad for 5 and 6 rings due to competing Claisen cyclisations. Use of Me3SiCl as a carbanion scavenger. So can use for easy access to small rings.
Can even make trans-fused cyclobutane to cyclohexane products:
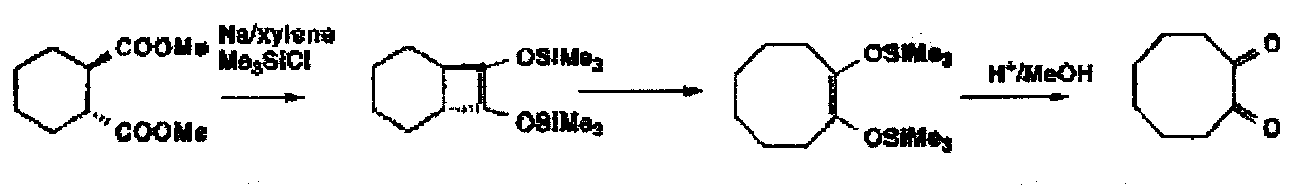
The initial product ring expanded by 2 by a 4e electrocyclic reaction to give the 8 membered diketone.
α,β-unsaturated ketones: a reaction to be careful with the protons:
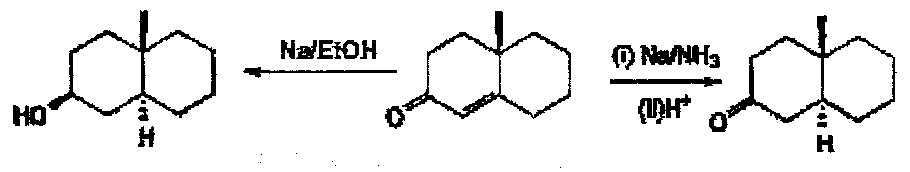
Stephen Reduction
Tin and HCl under heat will reduce nitriles to aldehydes effectively. Cl initially acts as a nucleophile, and then SnCl2 reduction proceeds. Water forms the aldehyde.
Choice of metal is important for all dissolved metal reductions, as is the availability of protons in the solvent.
Zn and Al are milder metals, and can be used to selectively remove O and S, e.g. in Clemmenson above which could be written as:
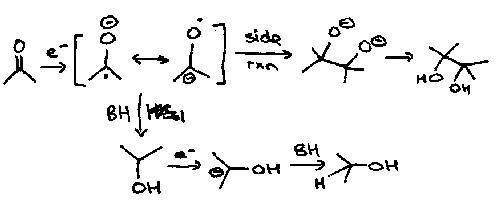
Carbon-Carbon Double and Triple Bonds
When they work, the reaction needs usually an amine/ammonia as solvent since alcohols would be too easily reduced.
Alkynes –

Catalytic reduction of alkynes give cis products, whereas sodium in liquid ammonia gives trans alkenes.
Conjugated C=C and the Birch Reduction
Isolated C=C are not usually reduced by dissolving metals, but in the case of conjugated dienes the resulting radical anion is accessible in amine solvents. This has been particularly applied to the reduction of aromatic compounds and the reaction is known as the Birch Reduction. The reagents are: the metal, the amine solvent and a proton source (EtOH).
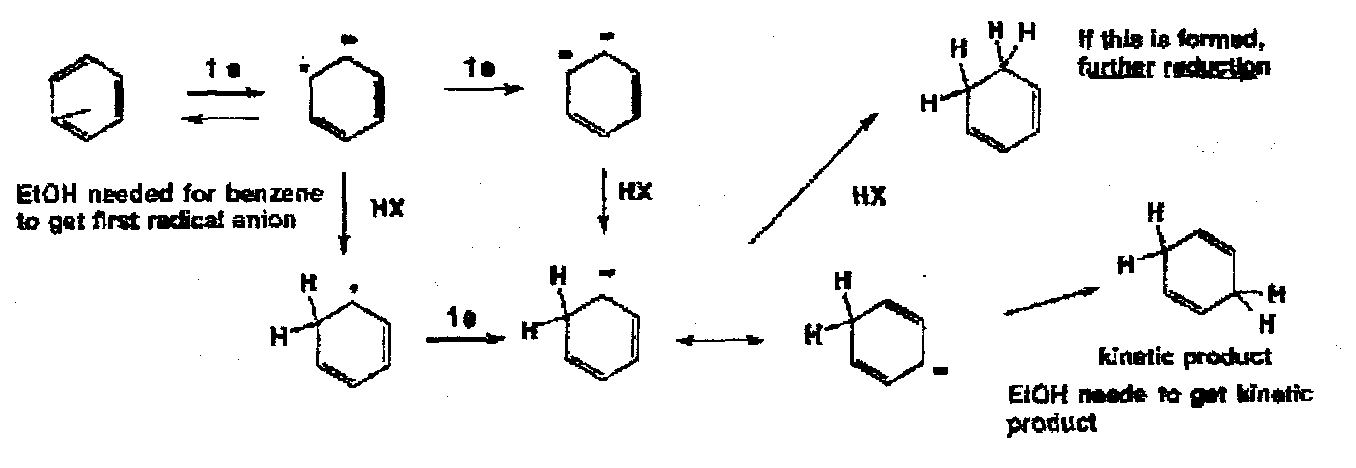
For the reduction of benzene itself, the ethanol has two roles: to provide an acid for easy protonation of the initially and unfavourably formed radical anion, and the provide kinetic and irreversible protonation of the final anion to give the unconjugated diene product. Again it is vital to appreciate the role of the proton donors in this type of reaction. Kinetic protonation of the pentadienyl anion always occurs in the middle rather than at the end, since the negative charge density is highest there. On the basis of this, it is quite easy to explain the extent of reduction of aromatic systems under different conditions.
In the case of anisole, you get more reduction than in the presence of EtOH.
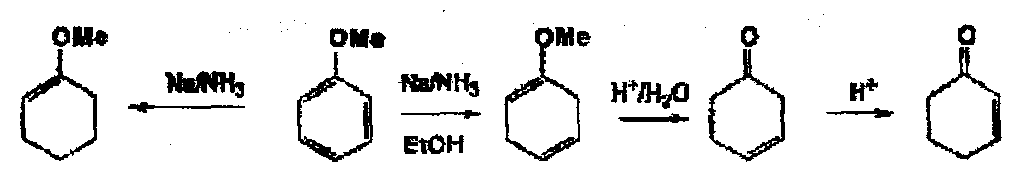
The orientation of the formation of the dihydro compound can sometimes be difficult to predict, but in general electron donating groups give 2,5 dihydro compounds where relevant withdrawing groups [COOH] give 1,4 reduction.
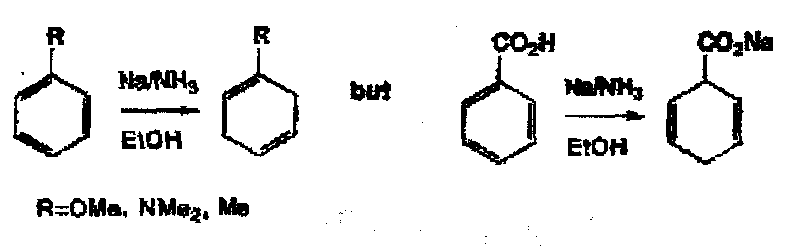
Reduction by Non-Metals
- Loss of Nitrogen as in the Wolff-Kischner and related reactions:
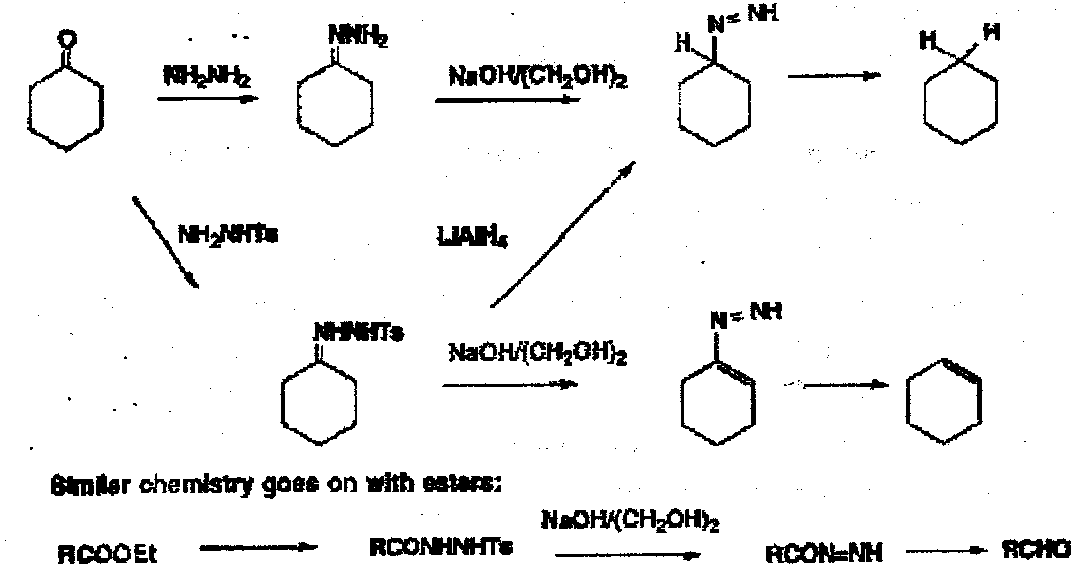
The loss of nitrogen in these reactions provides a good driving force. Anytime you get RN=NH this will fragment to give nitrogen and the product. Some of these reactions are done under very forcing conditions – boiling ethylene glycol with NaOH – so quite a bit of effort has been put into making the reactions easier.
Use of sulphonyl hydrazones; Bamford Stevens and Shapiro Reactions.
Beware of collapse of R-CH=CH-CH2-N=N-H by a retro-ene reaction.
- The Use of P(III), particularly in deoxygenation reactions and loss of sulphur.
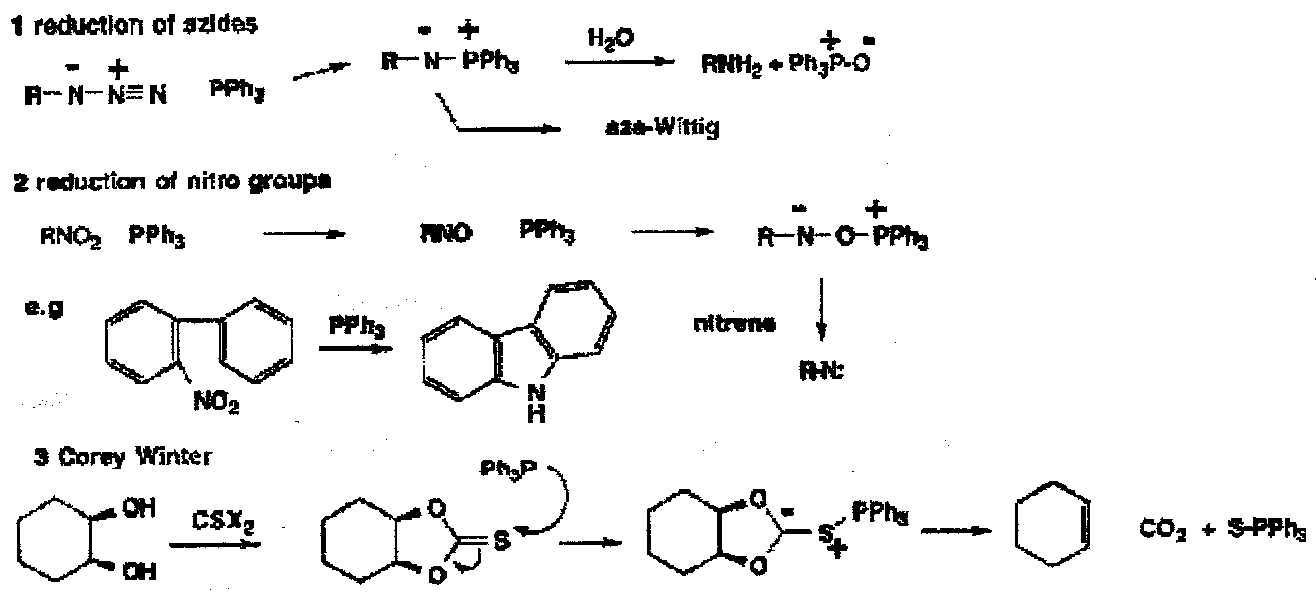
In these reactions, the initial step is nucleophilic attack by P(III) onto nitrogen, oxygen or sulphur, and the driving force for the reactions is the strength of the developing P=O or P=S.
The Corey-Winter and its later modifications relies on the gain of the P=S in the fragmentation of thionocarbonates.
Diimide
-O2C-N=N-CO2- in acid gives H-N=N-H. This then reacts via a pericylic mechanism to reduce alkenes. It attacks cis alkenes only, and will not attack α,β-unsaturated ketones.
Homogeneous Hydrogenation
As well as the dissolved metal reductions above, there are some other homogeneous reagents for reduction. They usually achieve homogeneity via soluble transition metal complexes. It minimises exchange / isomerisation.
An example is Wilkinson’s Catalyst, tris(triphenylphosphine)chlororhodium.
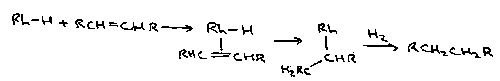
Stereoselectivity is dependent on how the metal complexes.
Another example is Evans’ Catalyst, [Rh(NBD)(disphos-4)]+BF4-. This hydrogenates, and is quite hindered, so selective.
Heterogeneous Hydrogenation
This occurs by using a catalyst and H2 (i.e. solid and gas). Various metals can be used, and they often show different selectivity:
|
Functional Group |
Converts To |
Catalyst Used |
|
C=C |
HC-CH |
Pd, Pt, Ni, Ru, Rh |
|
C≡C |
C=C |
Pd-CaCO3 with Pb (Lindlar) |
|
Aryl |
Cyclohexane |
Rh, Pt (low pressure), Ni, Pd (high pressure) |
|
C=O |
C-OH |
Pt, Ru (H+ cat, low p), Cu/Cr, Ni (high p) |
|
Ph-C(R)(O) |
Ph-CH2R |
Pd (H+ cat) |
|
Acyl chloride |
Alcohol |
Pd, Ni, Ru (difficult though) |
|
Ester |
Alcohol |
Cu/Cr, Ni |
|
Nitrile |
Amine |
Ni, Rh, with NH3 |
|
NO2 |
Amine |
Pd, Ni, Pt |
|
R-X |
R-H |
Pd |
|
Epoxides |
Alcohol |
Pt, Pd (H+ cat) |
The most common metals used as Pt and Ni.
Platinum is usually used when Nickel cannot be (since it is more potent, but more expensive). It will not attack carboxyl, esters or amides though. It is finely divided onto a carrier (usually activated C).
Nickel is usually activated as a porous, finely divided, Ni-Al alloy with NaOH (Raney). It often requires high temp and pressure. However it will work on most but chosen for aromatics and hydrogenolysis of S compounds. It is deactivated by carbon.
I am happy for them to be reproduced, but please include credit to this website if you're putting them somewhere public please!