Non Metal Chemistry
General discussion of the trends amongst the Non-Metals and reasons behind them, plus some comparisons between elements towards the end. Adapted from a variety of sources.
Non-Metal Chemistry Notes
General Properties of Non-Metal Elements
Non-Metal Characteristics
- Entropy – gases, volatile liquids and solids.
- Electrical Resistivity – typically insulators.
- Oxidizing Agents – electron acceptors.
- Molecular hydrides, oxides and halides.
- Acids / Bases – acidic oxides, but acidic, alkaline or neutral hydrides.
Ionisation Energies
General tendency for IE to increase across the periodic table. This is expected on the basis of effective nuclear charge, which increases across the period, so electrons are more tightly held and so harder to remove.
Ionisation Energy decreases down the Group, as principal quantum number increases faster than Zeff changes. IE Zeff2/n2. Zeff2 increases down the group, but not as fast as n2.
Must also consider distance from the nucleus, i.e. penetration and shielding effects (cf. O(2p4) and N(2p3).
Comparing Group 13 trends, we note that the value for Ga is slightly greater than that for Al, which is not predicted. This is due to the full d-shell which inadequately shield, so Zeff rises. The same effect is noted on moving from In to Tl, where this time a full 4f shell contributes to Zeff due to its poorer shielding.
Lower Periods: Ionisation Energies rise more slowly with charge, so higher oxidation states are predicted to be more favourable.
Electron Affinities
This is the reverse of ionisation (energy change when an electron is added to an atom/ion.
It is seen that electron affinities generally increase on crossing the periods (same trend as for IE’s, and for the same reason). Again, the trend is interrupted at Group 15 due to the np3 configuration (spin pairing must occur).
d-block and f-block contraction is also evident, such that 3p/4p and 5p/6p have similar values.
Finally, we note that the EA’s for the first row elements are generally lower than those for the 2nd row elements, particularly for Groups 15, 16, 17. This is a result of the very small sizes of the atoms N, O, F (charge-to-size ratio high, i.e. electron repulsion higher).
Covalent Radii
Decrease in covalent radius across the periods. This is due to Zeff increasing (contraction of orbitals). Also, radii increase down the groups. This results from the population of shells with higher principal quantum number (further from the nucleus). Also note the similarity between 3p/4p and 5p/6p elements due to d/f-contraction again.
There is less decrease across 2nd and lower periods compared to the 1st. Approximately 50% increase in covalent radii from 1st to 2nd row. Coordination numbers are thus likely to be higher (1st row CN ~4, while 2nd row up to 6).
Electronegativities
Increases regularly across the periods, with the rate of increase tending to decrease as the p-block is descended. Also tends to decrease down the group, indicating a transition to more metal-like properties, and a decline in ionic character.
Again, we note the similarity of the 3p and 4p elements, and often the 4p value exceeds the 3p, against the trend (Ga > Al, Ge > Si).
Must remember though that typical electronegativity scales are for the atoms, and so they will vary with e.g. Oxidation State (e.g. TlIII > TlI).
Why Not Ionic Structures?
- Contraction upon ionisation not large.
- Lattice energy small.
- Covalent bonds sufficiently strong (contrast metals).
- Ionic structures could be stable with respect to the elements but covalent structures even more stable (due to large radius of non-metal cations).
- For most metals, covalent bonds not sufficiently strong to cause collapse of ionic structures, just covalent character in the bonding.
c.f. CaCl(s) → Ca(s) + CaCl2(s) ΔH<<0
Relativistic Effects
One of the consequences of special relativity is that objects moving close to the speed of light increase in mass. The importance of this to chemistry is that for some of the electrons in the heavier elements, velocities are an appreciable fraction of the speed of light as a result of high nuclear charge, and are therefore subject to these effects.
As the velocity, and hence the mass, of the electron increases, the effective size of the orbital decreases and its energy is lowered. The effect is most pronounced for electrons in s orbitals, since it is these orbitals which have the greatest density near the nucleus, rather less marked for p orbitals and relatively unimportant for d and f orbitals. For the heavier p-block elements Tl through to Rn there is an appreciable stabilisation of the electrons in the 6s orbital which has important consequences.
Properties of the Elements
Group 13
Only non-metal in this group is Boron, which is classed as a metalloid. Elemental boron has many polymorphs, each of which is a polymeric or macromolecular solid. Most of these contain B12 icosahedra linked in complicated 3D arrangements. The structural complexity is a manifestation of the electron-deficient nature of Boron.
Al and Tl have typical close-packed metallic structures with 12 nearest neighbours. In has a slightly distorted close-packed structure with 4 atoms marginally closer than the other 8, but Ga has a rather unusual structure in which each atom has one neighbour considerably nearer than the others which occur in pairs. In this sense it is similar to solid iodine (Ga2 units).
Electronegativity (and the Alternation Effect) can be used to rationalise the above. B is too electronegative to be a metal, but the least electronegative elements (Al, In and Tl) adopt typical metallic structures. Ga, the most electronegative element of the group, has a structure in which incipient bond localisation typical of the metalloids is present.
Group 14
Carbon is non-metallic, and usually found as graphite or diamond, the structures of which are well-known. Extensive π-bonding is a feature in the structures of the newly discovered molecular forms of carbon (the fullerenes).
Si and Ge both have cubic diamond structure and are classified as metalloids largely as a result of semiconducting properties. These are elements of intermediate electronegativity in which localised covalent bonding occurs but for which the valence s-p energy separations are small enough to result in fairly closely spaced bands.
Sn lies close the metalloid-metal border, and exists as two allotropes (grey and white tin). The former has the diamond structure, while the latter is much less regular with 4 nearest neighbours, but with additional atoms much closer than the second nearest neighbours in grey (diamond-like) tin structure. It is suggested that this reflects the two oxidation states of tin, Sn(IV) in grey-tin, while Sn(II) in white-tin (more strictly speaking, this reflects the number of electrons being used in bonding (4 vs. 2) as opposed to Oxidation State, which is clearly zero for both). These is seen on dissolving the respective allotropes in aqueous HCl.
Lead has a typical close-packed metal structure, with particularly long Pb-Pb distances indicative of the presence of a non-bonding 6s pair of electrons.
Group 15
Nitrogen is a diatomic gas, while P does not exist as P2 molecules except under conditions of high temperature and low pressure in the gas phase. The stable forms instead are white phosphorus (tetrahedral P4 molecules) and a variety of polymeric allotropes such as red, black and violet phosphorus. They contain three coordinate phosphorus in which each P atom is linked to 3 others by single P-P bonds. This reflects an important difference between the chemistry of P and N, and between the 1st and 2nd row elements in general, which is the reluctance of the heavier elements to form stable π bonds and the corresponding prevalence of element-element single bonding (or catenation, see below).
The common α forms of As, Sb and Bi are isostructural and consist of hexagonal sheets similar to black P. other allotropic forms exist. Down the group there is a tendency for the ratio of long to short bonds to decrease, such that the bonds become more nearly equal in length. This is a definite trend from localised covalent bonding in a sheet structure to a more delocalised, 3D metallic bonding (consistent with decreasing electronegativity of the elements).
Group 16
O2 and O3 known, while S2 and S3 not except at high temperatures. S-S bonded species much more prevalent, the number of allotropes of which is extremely large. S8 rings are the most common, but there are many other sized rings, as well as polymeric, fibrous forms which contain helical chains of S atoms.
Se has properties consistent with a non-metal, and its allotropy is extensive, although not nearly as much so as Sulphur. Se8 rings and a hexagonal form of unbranched helical chains are favoured.
Te, by contrast, exists as only a single allotrope which is isostructural with hexagonal Se. A similar trend in bond distances to that noted for the heavier Group 15 elements is also apparent.
Polonium is the least electronegative and so most metallic in character. It has a simple cubic (Primitive) structure, and is the only element for which this is found.
Group 17
The halogens exist as diatomic molecules under normal conditions, with F2 and Cl2 as gases, Br2 a liquid and I2 a solid. Low temperature solid Cl2 and Br2 are isostructural with I2 (layered). The F2(s) structure is also layered, but differently. It is worth noting that the I2 bond length is longer in the solid than the gas, and this is a result of the closeness of the intermolecular contacts, a feature common in iodine chemistry.
Binding Energies
The binding energy (or atomisation energy) trends:
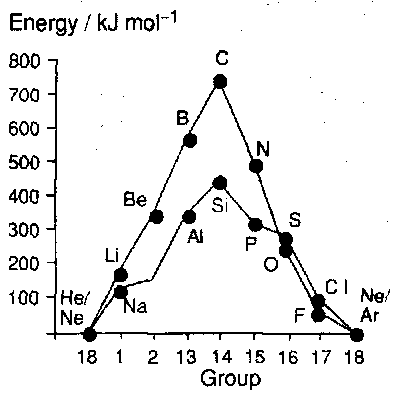

General Trends for Compounds
Oxidation States, Valence and the Inert Pair Effect
General trend seen that Oxidation States of N or N-2 are found. General exceptions include compounds with unpaired electrons (e.g. NO, NO2, ClO2). Radicals are not frequently found amongst the p-block however, as the s-p hybrid orbitals that usually form in these compounds have fairly good directional overlap, so reasonably strong bonds are formed such that any radical species tend to dimerise, unless the electron is extensively delocalised.
The N Oxidation State can largely be predicted based on Lewis’ Octet Rule. The N-2 Oxidation State arises due to an effect known as the Inert Pair Effect. For example, in Group 13 the +3 oxidation state can be seen as the removal of 3 electrons (ns2np1) leaving an inert noble gas core, whereas the +1 state involves the removal of only the np1 electron. The N-2 State is common for the heavier elements, particularly so for Tl in Group 13.
A standard explanation is that the relatively large effective nuclear charge of thallium, resulting from the presence of the 4f and 5d elements, coupled with relativistic effects, leads to substantial stabilisation of the 6s pair relative to the 6p electron, and it is this factor which gives rise to the term inert pair itself. However, this is only part of the explanation. Although the 6s pair is stabilised by these effects, it can hardly be considered to be too low in energy (i.e. core-like) in view of the relevant ionisation energies which are comparable with those of the lighter elements.
Realistically, the effect is a combination of two factors. Firstly, the promotion energy involved in achieving the valence state is high, but an equally important point is the weak bond formed by this element as a result of its large size. Thus, for boron, the energy required to involve the 2s pair in bonding (the promotion energy) is more than offset by the formation of three strong covalent bonds and boron is therefore more likely to occur with an oxidation state of +3. In the case of thallium, the opposite trend is true and the formation of two extra, much weaker bonds is not enough to compensate for the considerable promotion energy.
This idea extends through to all the p-block groups, including the noble gases.
Bond Energies
- Better energy match means stronger, shorter bonds.
- Overlap, fractional population of bonding electrons under the influence of bonding nucleus; better overlap, stronger bond.
- Core electron repulsion (important for O, F, 2nd row down).
- Non-bond electron repulsion (important at small bond distances).
Homopolar Single Bonds
- Absolute bond strength values are not important, more important to establish trends and reasons.
- H-H strongest; no core electron repulsion.
- Down group IV(14), bond strength goes down as overlap and core repulsion goes up.
- C-C > N-N > O-O, F-F
- F-F << Cl-Cl non-bonded electron repulsion important.
- 2nd row onwards, across period, irregular, complex reasons.
Homopolar Multiple Bonds
- 1st row, multiple bonds strong, good overlap
- However, note N=N < O=O due to non-bonded electron repulsion
- Down group, poorer overlap because radius is higher and core electron repulsion increases if radius drops to improve overlap
Heteropolar Single Bonds
Element-Hydrogen Bonds
- Across period, B(E-H) increases, no core electron repulsion, so overlap can increase as covalent radius can be small
- Down group, B(E-H) decreases, as overlap decreases, as energy match condition is worse
Element-Fluorine Bonds
- Across period decrease, core electron repulsion increases as atoms become smaller
- Down a group, should decrease as core electron repulsion increase and overlap decreases:
C-F > N-F > O-F > F-F
order due to non-bonded electron repulsion
- C-F vs. Si-F dπ-pπ bonding in Si-F
- N-F vs. P-F dπ-pπ + reduced non- bonded electron repulsion
- O-F vs. S-F dπ-pπ bonding
Element-Halogen Single Bonds
- The trend is always E-F > E-Cl > E-Br > E-I (true for nearly all elements), except: F-F and Cl-F due to large non-bonded electron repulsion in F-F bond
Heteropolar Multiple Bonds
- Trends similar to homopolar bond energies
- Down group, Bond energy decrease as overlap decreases
- Across period, Bond energy decreases as core repulsion increases
- (E=O) > (E=S), overlap and core repulsion
- (E=O) > (E=N), due to overlap considerations
Changes with Oxidation State
For all cases B(E-Hal) decrease as oxidation state increases.
- Main reason, contracted central atom orbitals, overlap improves but core electron repulsion increases.
- B(E-F) decreases least as oxidation state increases, therefore highest oxidation state most likely with F as ligands.
Comparison with d and f Blocks
We have seen that bond strengths generally decreases down p-block Groups, but the opposite is often true with d-block groups, with third row TM’s tending to form the strongest bonds. We also note that the higher Oxidation States tend to be more stable for the 3rd row TM’s as well, whereas down the p-block Groups high Oxidation States become less stable. This in fact is the reason for the bond strength trends, as the relativistic stabilisation and contraction of the s and p orbitals for the third row elements leaves the d-orbitals more exposed and so overlap is better and bonds are stronger. A similar situation is found for the f-block elements where covalent bonding is much more prevalent in the chemistry of the actinides.
Ketelaar’s Triangle
The types of compounds formed by p-block elements can largely be rationalised by electronegativity differences. Large Δχ tend to be ionic, those with intermediate Δχ polymeric or macromolecular structures. For compounds with a small Δχ, the type of compound depends on whether the elements have a low or high electronegativity. In the former case, the compounds are likely to be metallic for much the same reason that elements with low electronegativity are metals. In the latter case, we expect relatively non-polar molecular species.
A useful way to visualise these ideas is with Ketelaar’s Triangle:

Consider the covalent-metallic edge. In both types of bonding it is the overlap of orbitals which is important and the elements themselves provide a good example of the trend from one form to the other, e.g. Li & Be are metallic, while B & C are metalloid, whereas N2, O2 & F2 are covalent. Clearly the differences in electronegativity are always zero, so the changes that occur reflect the change in the magnitude of the electronegativity.
The ionic-covalent edge is characterised by decreasing electronegativity difference, which is well illustrated by the fluorides of the elements. LiF and MgF2 are highly ionic, whereas PF5, SF6 are covalent. Intermediate compounds such as BeF2 are polymeric or macromolecular solids.
The metallic-ionic edge is also characterised by increasing electronegativity difference and can be illustrated with the compounds of lithium, e.g. Li (metallic), Li3As (metallic alloy), Li3N (polymeric), Li2O (ionic).
Compound Formation
The more bonds formed, the weaker those bonds tend to be. For example:
|
SF2 |
SH2 |
|
SF4 |
SH4 |
|
SF6 |
SH6 |

As with more complete MO pictures, 3c4e bonding models but substantial electronic charge on the terminal atoms. This can be represented in simple form by valence structures with ionic contributions, with the central atom having no more than four electron pairs in any structure, e.g.
F-Xe+ F- ↔ F- Xe+-F
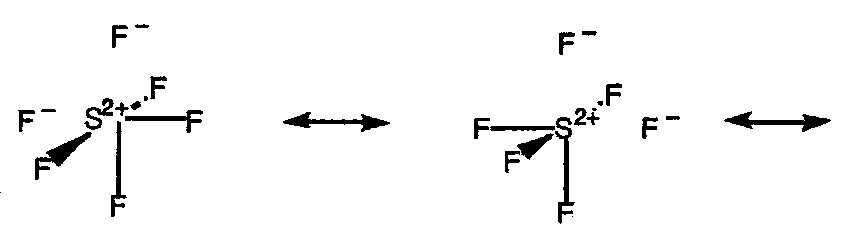
Reasons for formation of hypervalent compounds in the 3rd and lower periods:
The major contributions do not come from the ability to use valence d orbitals, but from:
- Larger atomic size which allows higher coordination number without excessive steric crowding;
- Lower electronegativity which accommodates more easily the positive charge required without d orbital contributions. This also explains why hypervalent compounds are more often found with highly electronegative terminal atoms.
Variation of A-B Bond Enthalpy with n in ABn:
Progressive decline in bond strength with increasing n has implications for the stability of hypervalent compounds.
e.g. instability of SH4 and SH6 is partly explicable if the S-H bond behaves in the same way as the S-F bond shown above.
Bonding Models for Hypervalent Compounds
Simple Lewis or valence pictures are useful for representing bonding and non-bonding electrons.
They may be used in conjunction with VSEPR to predict shapes (although not always reliably: [SeCl6]2- is a regular octahedron in spite of having one non-bonding electron pair).
Simple directed valence models interpret the number and geometry of electron pairs around an atom using hybrids with d orbital contributions. E.g. sp3d for five pairs in a TBP geometry, sp3d2 for six octahedral pairs. Details can be found in books but it is now recognised that such models seriously overestimate the d orbital contribution to the bonding.
MO Diagrams show the pattern of bonding orbitals predicted with or without d orbital contribution, and show that such contributions are not essential. Accurate MO calculations show that d orbitals in fact make very little contribution.
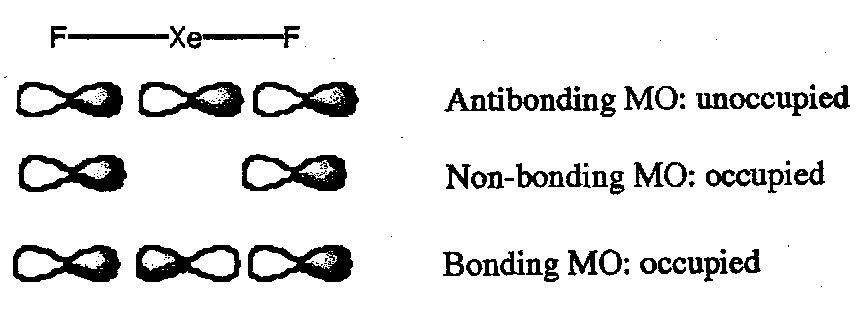
3c4e bonding models give simplified MO pictures based on the assumption that only p orbitals contribute to bonding, e.g. application to XeF2:
When trying to relate valence electron count and steric number to a selection of molecules and ions, it is found that:
- There are a wide range of isoelectronic analogues, but same valence electron count does not imply similar properties.
- Monomeric multiply-bonded structures with C, N and O often replaced by polymeric structures in lower rows.
- Steric Number > 4 requires octet expansion (hypervalence) and is not found with elements B to F.
Variation of Bond Enthalpy with Bond Order
Compounds with multiple bonds between C, N and O are very common.
Multiple bonds between heavier non-metals much less stable.
e.g.
- Contrast elemental structures N2 and O2 with P4 and S8.
- R2C=CR2 → (CR2)n requires catalyst, but R2Si=SiR2 → (SiR2)n unavoidable unless R is very bulky.
- CaC2 contains discrete C22- ions, while CaSi2 has polyermic Si- layers with each Si forming 3 single bonds as in elemental As.
- Contrast CO2 with (SiO2)∞.
Size and Polarisability Trends
Covalent and Anion Radius increases down group, as does polarisability.
Note characteristic structural differences in solids as a result: MO, MO2, MF2 vs. MS, MCl2, MS2 etc.
A-B Single Bond Enthalpies
Trends:
- C-X > Si-X > Ge-X for X = H or C.
- C-X < Si-X > Ge-X for X = O, F, Cl.
- Single bonds between elements N, O, F are “anomalously weak” – due to lone pair repulsion. As size increases, repulsion decreases, so bond gets stronger.
- Alternation Effect apparent in N-Cl < P-Cl > As-Cl < Sb-Cl.
Compounds of the p-block Elements
Halides
The fluorides of the s & p-block elements in their Group Oxidation State show a tendency to move from ionic compounds on the left (e.g. MgF2) via polymeric species (e.g. AlF3) to covalent compounds such as PF5. This can be explained using Ketelaar’s Triangle as described earlier (i.e. the difference in electronegativity of the two elements forming the compound).
F is the most electronegative element. Thus, in forming complexes with electropositive elements on the left hand side, particularly those near the bottom left, there will be a considerable degree of charge transfer resulting in the formation of the negatively charged F- ion, and a positively charged element cation E+. Hence these compounds will tend to be ionic, with predominantly electrostatic bonding.
In contrast, elements to the top right of the p-block have a small electronegativity difference, so charge transfer from F is small and the compounds are relatively non-polar, with covalent bonds. They are typically gases or low boiling point liquids as a result.
Intermediate compounds such as SbF5 form covalently bonded monomers, but in the solid / liquid phase they interact strongly to form polymeric species due to the highly polarised covalent bonds that have formed. SbF5 forms a linear polymer in the liquid state, and tetramers in the solid state.
The lower oxidation states tend to show similar trends. For example, in Group 13, B2F4 is covalent and TlF is ionic. Similarly, GeF2 is polymeric (unlike GeF4 which is covalent) which can again be rationalised by electronegativity differences (Ge(IV) centre will be more polarising than the lower oxidation state).
It is important, however, to not ignore other factors that contribute. In particular, element size and the nature of the vacant orbitals which are present. Thus, GeF2 is also more likely to be polymeric than GeF4, since the former is only two-coordinate, and Ge(II) is larger (can expand its coordination number more).
As expected, for the other halides there are fewer ionic compounds, reflecting the decreased electronegativity of the other halides. There is also a trend to intermetallic or alloy properties, particularly with iodide.
Oxides
Continuing the theme from the halides, the high electronegativity of O allows for a range of ionic through to covalent compounds moving from left to right across the periodic table.
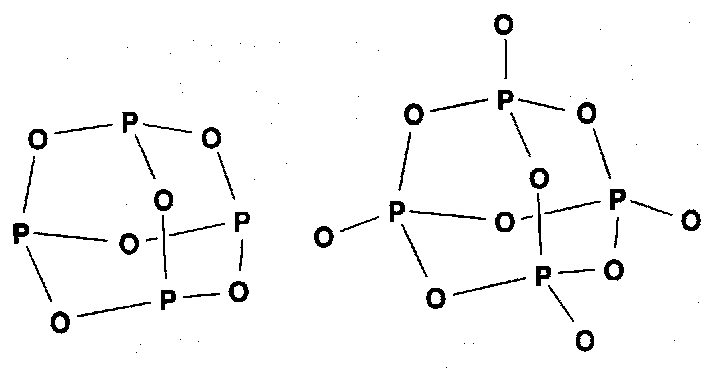
The difference in chemistry between the 1st row and the lower rows is highlighted by considering the multiple bond strengths discussed earlier. Thus, molecular compounds such as CO2 and the various Nitrogen Oxides form due to favourable π-bonding interactions, whereas Si and P oxides tend to catenate far more, giving rise to structures such as:
Hydrides
There is a greater tendency for covalent compounds here, reflecting the low electronegativity of H. Polymeric species are rare too, as BeH2, BH3, AlH3 and GaH3 being the only ones. From Group 14 onwards molecular covalent species are formed.
Catenation and Polymerisation
Catenation
Chains and rings with E-E bonds. Note bond strength trends:
C-C > Si-Si > Ge-Ge, but N-N < P-P and O-O < S-S.
Silicon
Silanes SinH2n+2 known with n = 1-8, but much more reactive than alkanes. Fluorosilanes SinF2n+2 up to at least n = 16 can be prepared.
Many metal silicides contain Si-Si bonds, e.g. CaSi2.
Phosphorus
Polyphosphanes PnHn form rings; PnH2n+2 and PnHn-4 are also known.
Many metal phosphides contain P-P bonds, e.g. SrP2.
Sulphur
Strong tendency to catenate. Has many allotropes, containing Sn rings (n = 6-20) and S∞ chains.
Many polysulphides, polysulphanes and halides.
Note also the importance of the disulphide S-S bond in protein structure.
Multiple Bonds
Generally much harder to make with Si and P than with C and N.
Preparative reaction:
SiCl4 + 2RMgBr → R2SiCl2 [+ 4Li+C10H8-] → R2Si=SiR2 → collapses to (SiR2)n unless R is bulky.
Similarly for RP=PR.
Analogous compounds with heavier group 14 elements are often non-planar.
S=S bonds are more common, e.g. in S2O32- and in S=SF2 (more stable than FS-SF).
Polymerisation
Silicates
- Form about 95% by mass of the minerals in the earth’s crust and mantle.
- Orthosilicates, e.g. Mg2SiO4 contain discrete tetrahedral [SiO44-] ions.
- Disiliates, e.g. Sc2Si2O7 contain corner-sharing pairs of tetrahedra {Si2O76-].
- Metasilicates contain [SiO32-]n rings (Be3Al2{Si6O18}) or chains (CaSiO3) of corner sharing tetrahedra: note contrast with carbonates (discrete planar CO32-).
- Further polymerisation gives double chains, layers and 3d frameworks.
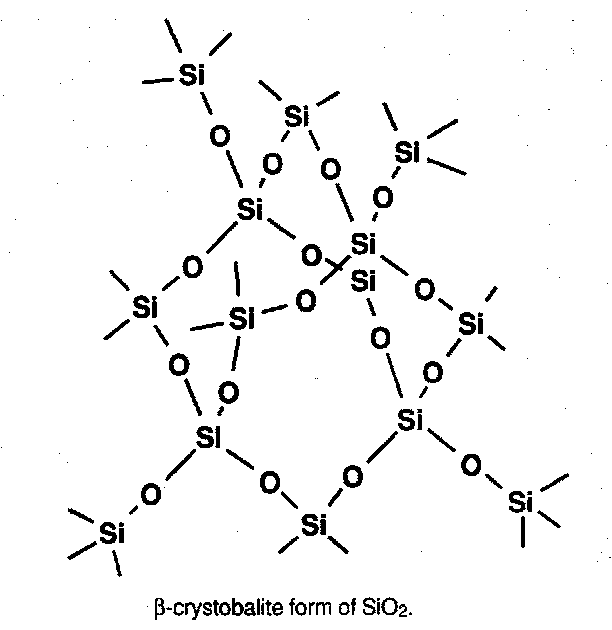
- SiO2 (many polymorphs) has SiO4 tetrahedra sharing all corners.
- In aluminosilicates some tetrahedral Si is replaced by Al, the lower nuclear charge being compensated by addition cations, e.g. NaAlSi3O8.
- Zeolites are framework aluminosilicates with microporous structures. During synthesis the pores are filled with hydrated cations, but these may be removed and replaced with H+. Zeolites are important shape-selective catalysts used for cracking and isomerisation of hydrocarbons in petrochemicals processing.
Phosphonitrilic Compounds
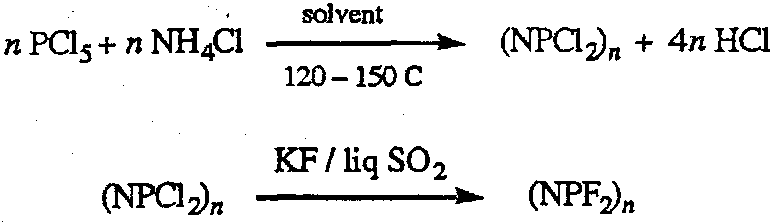
Cyclic oligomers with n = 3 and 4 predominate in normal preparation, but higher polymers can be made on controlled heating.
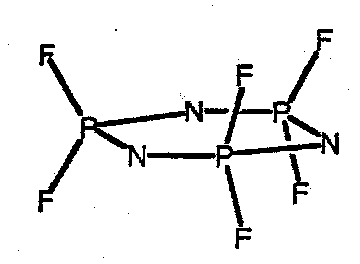
(NPF2)3 has a planar skeleton but most rings are non-planar. However all N-P distances are equal with P-N-P angles close to 120o. Bonding is hard to describe simple terms, but note isoelectronic analogy with silicones.

Sulphur Nitrides
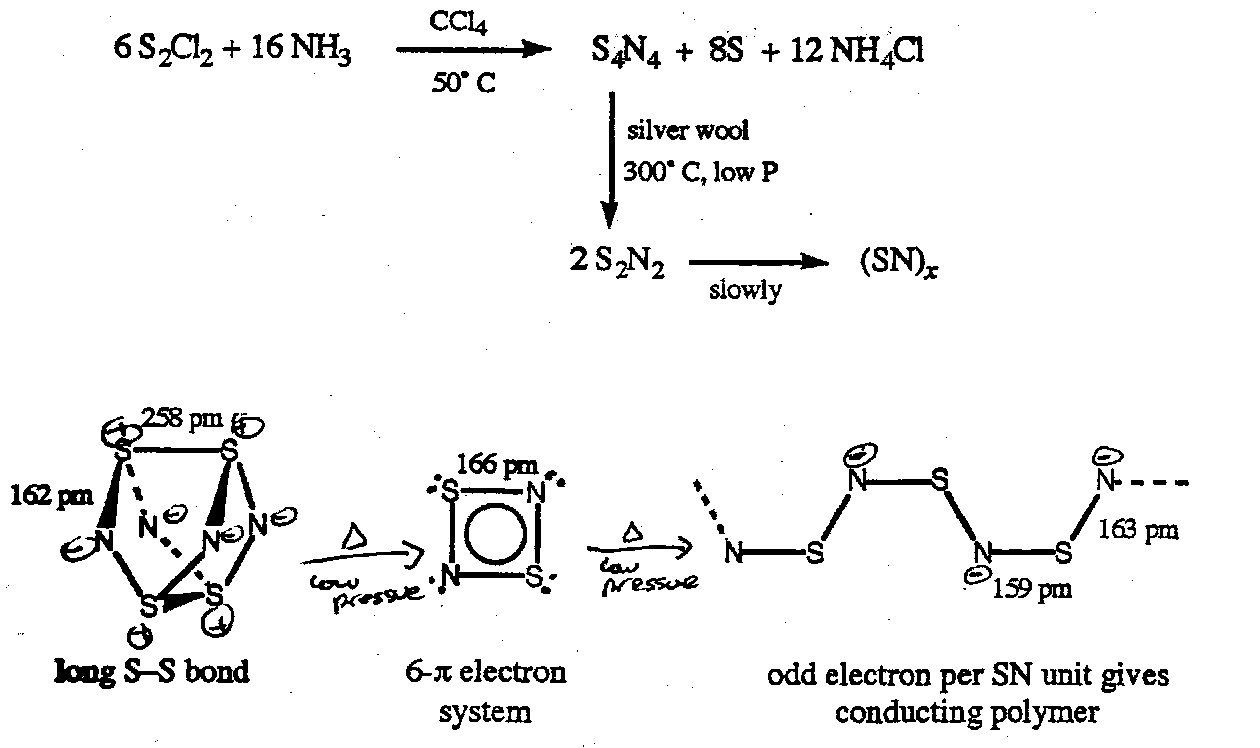
Zintl Principle
Helpful for identifying structural similarities between compounds. Another name for this idea is the isoelectronic principle, which is much more descriptive, since it indicates that the key to structural similarity is to be found in the number of (valence) electrons.
For example, consider the polymeric compounds with the cubic diamond structure, e.g. III-V compounds such as Boron Nitride, Aluminium Phosphide and Gallium Arsenide, and the II-VI compounds Zinc Selenide and Cadmium Telluride, and also the I-VII compounds Copper(I) Bromide and Silver Iodide.
For more ionic systems, an example is the structure of MgB2, with each Boron present as B- (isoelectronic with carbon) in planar hexagonal sheets just like graphite. Similarly, LiAs is isoelectronic with grey Se, whilst CaSi2 has Si- isostructural with black phosphorus.
Clusters, especially Boranes
Classification of Main-Group Clusters
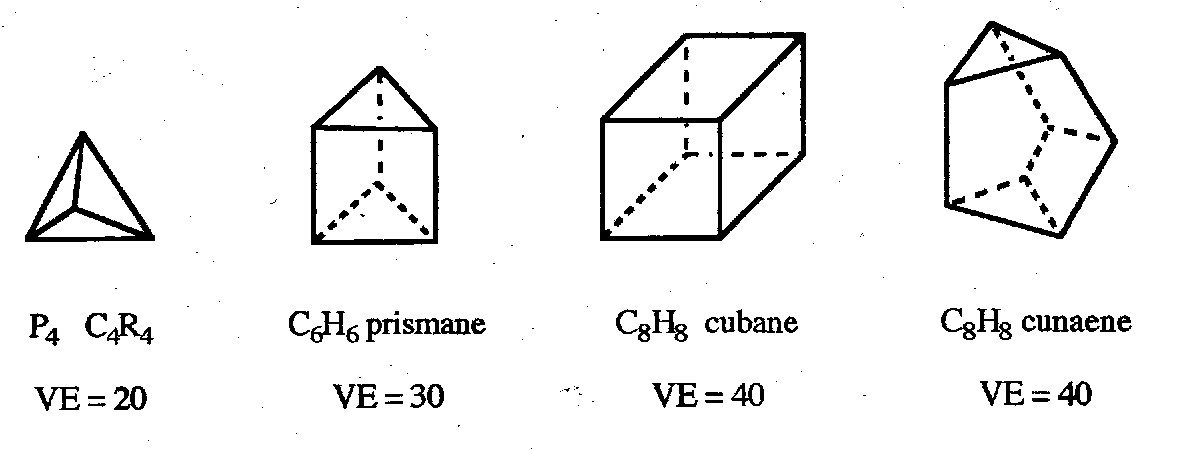
Electron-Precise Clusters
Three-connected clusters with just the right number of valence electrons to form 2c2e bonds. Simple examples with n atoms have 5n valence electrons.
Electron-Rich Clusters
Have valence electrons > 5n. Additional electrons would occupy antibonding MOs in the electron-precise structure. This leads to bonding-breaking or (less often) bond-lengthening.
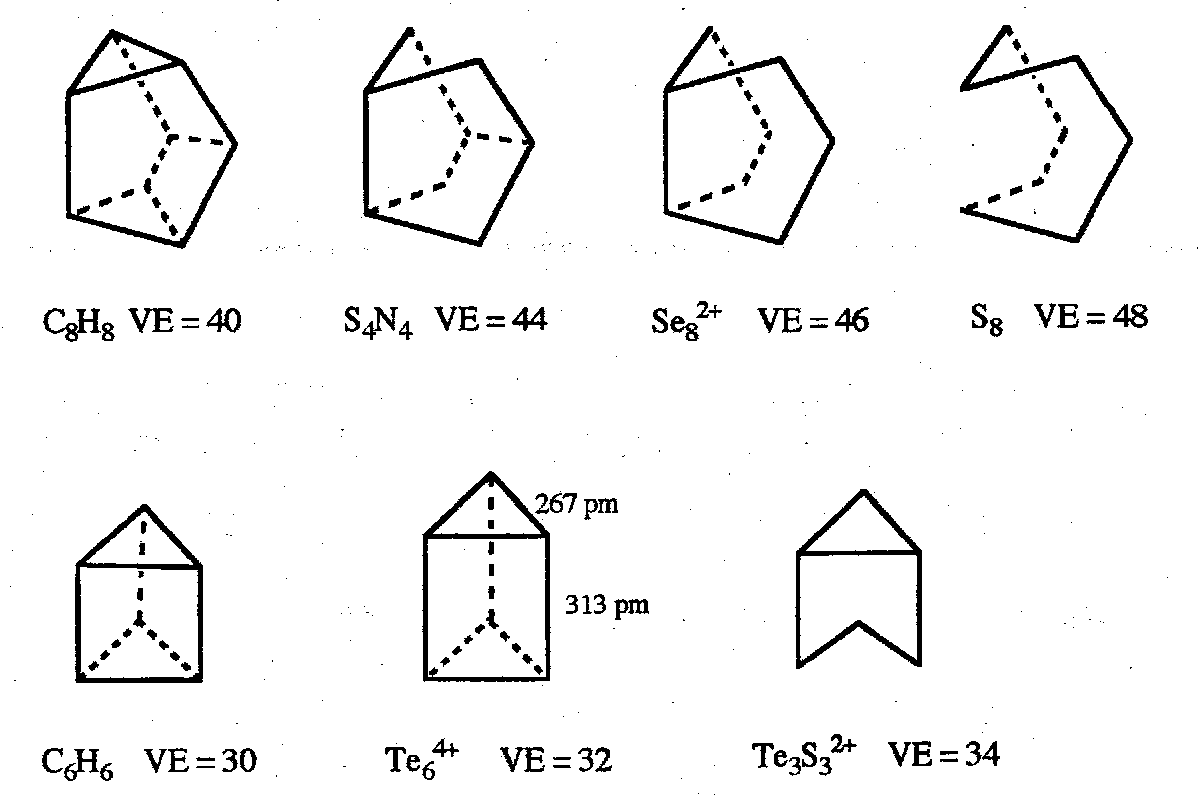
Electron-Deficient Clusters
Clusters with insufficient electrons for 2c2e bonding description are sometimes described by this, although electron-delocalised is a better term.
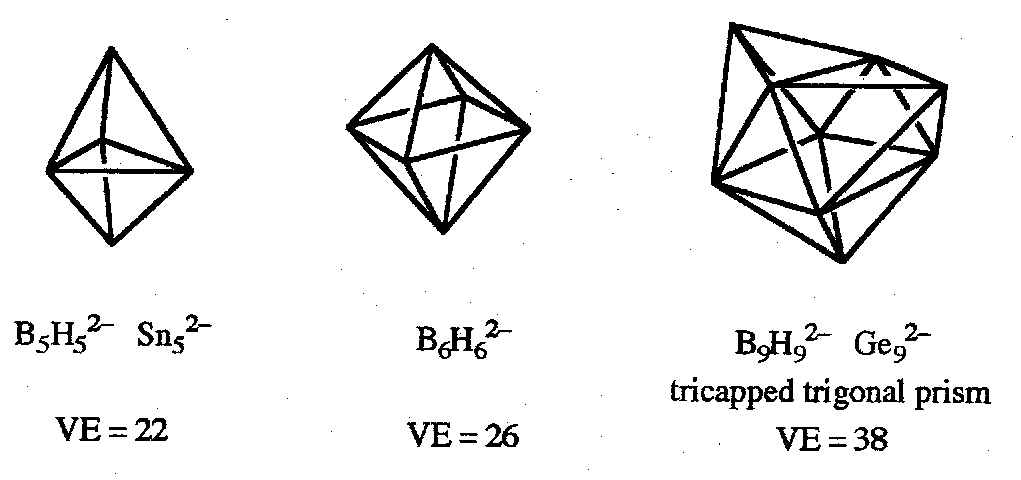
Structures are often based on polyhedra with only triangular faces (known as deltahedra) which maximise the bond connectivity.
Borane Structures
The closo-/nido-/arachno- classification is exemplified by the structures below:

Closo-boranes have n-atom boron cages forming a deltahedron with n vertices. The major series are anions [BnHn]2- with n=5-12.
Nido-boranes (“nest-like”) have n boron atoms roughly at positions based on an n+1 vertex deltahedron. General formula BnHn+4, e.g. B5H9 (based on octahedron).
Arachno-boranes (“web-like”) have n boron atoms at positions based on n+2 vertex deltahedron. General formula BnHn+6, e.g. B4H10 (based on octahedron).
Bonding Boranes
Can sometimes be understood in terms of localised 3c2e bonds, e.g. B4H10 – valence electron count = 22. Six terminal B-H bonds use 12 electrons; 2 form a 2-center B-B bond, leaving 8. Each of the four bridging H’s are then involved in 3-centre B-H-B bonds.
Larger clusters cannot be easily understood in these terms.
Bonding in B6H62-
We can use our knowledge of octahedral SALCs to construct a more detailed picture for the anion B6H62-. The SALCs formed are identical to those formed by the σ and π ligand orbitals in an octahedral complex, but in the cluster there is no central atom. Bonding is only between the B-H units themselves.
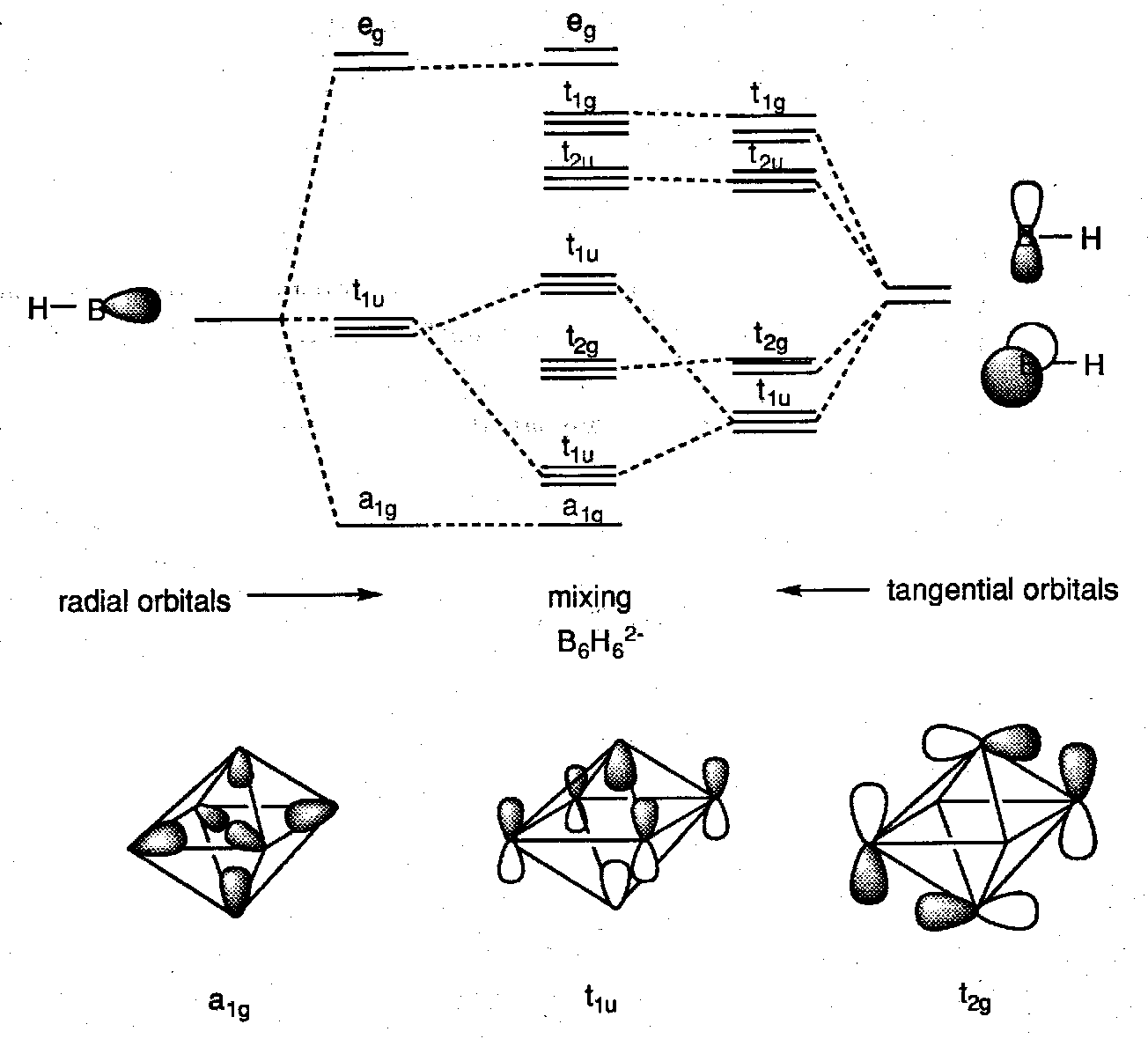
Delocalised MO models are therefore required. Wade’s Rules, which generally apply to boranes with n ≥ 5 are based on the prediction that an n-vertex deltahedron should have n+1 skeletal bonding MOs.
Closo-boranes therefore have 2n+2 skeletal bonding electrons, or total VE = 4n+2, e.g. [B6H6]2- (VE = 26), [B12H12]2- (VE = 50).
We take as our basic building block in forming these clusters the B-H unit. It has fragment orbitals of the A-H type:
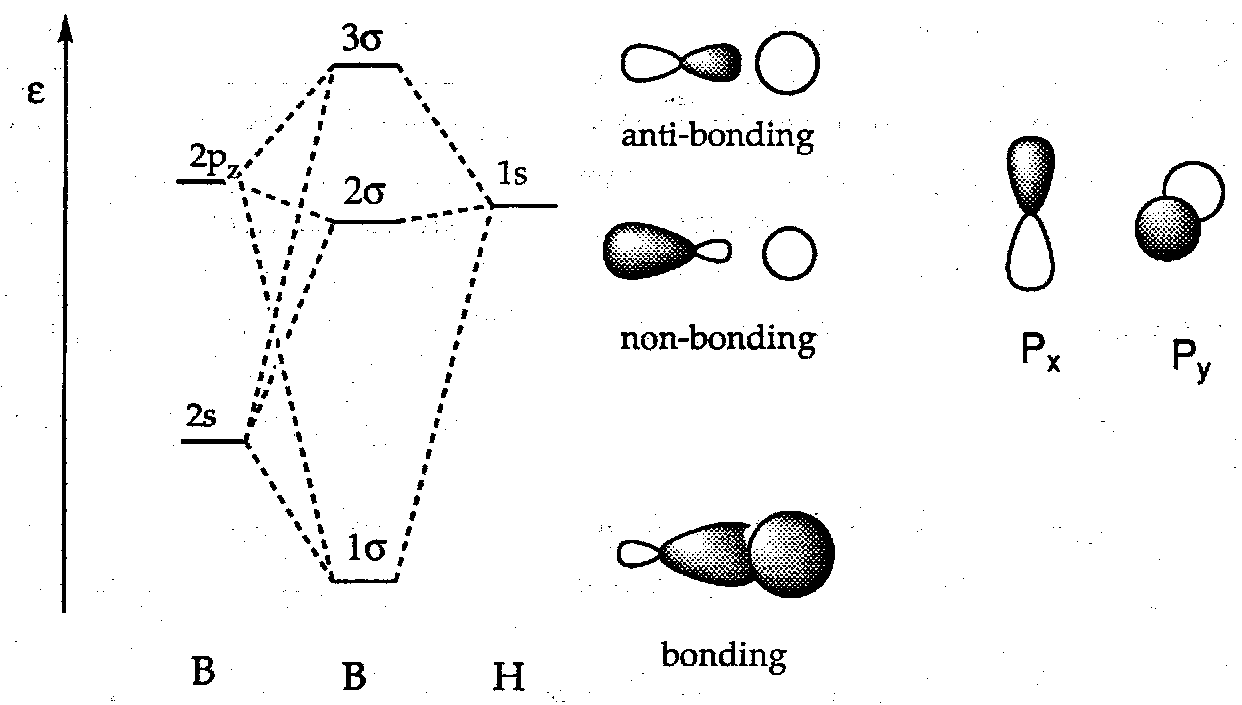
The B-H bond is a normal 2 electron bond. To a first approximation we can treat these separately from the rest of the cluster.
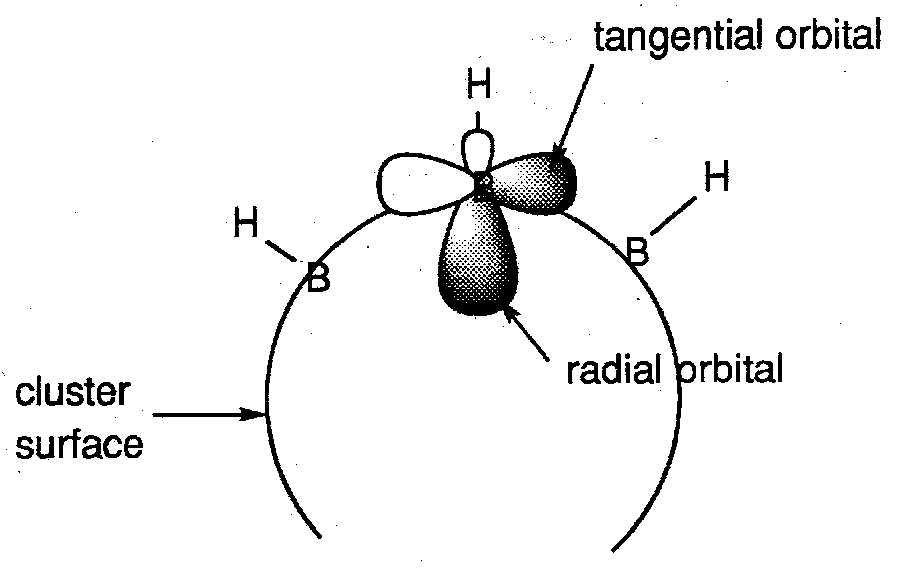
We can treat the cluster as an almost spherical surface of B-H units aligned along radii of the sphere.
Starting from closo-[Bn+1Hn+1]2- removing one BH2+ and adding 4H+ generates nido-[BnHn+4]; this does not change the number of skeletal bonding electrons, and therefore nido-boranes BnHn+4 have 2n+4 skeletal bonding electrons.
Arachno-BnHn+6 can similarly be generated from nido-Bn+1Hn+5, giving 2n+6 skeletal bonding electrons.
Wades Rules often (but not always) work for p-block polyanions, e.g.
Sn52-, VE = 22. Assume each Sn has one lone pair, so 12 = 2n+2 skeletal electrons appropriate to close structure observed.
Sn94-, VE = 40. So 22 = 2n+4 skeletal electrons, nido-structure (capped square antiprism).
Two predictions of Wade’s Rules applied to boranes are:
- Isoelectronic replacements do not change the structure. Carboranes are based on the replacement of C-H for B-H-. Transition metals and organometallic fragments may also be incorporated giving metalloboranes. Appropriate modifications of the electron counting rules is needed.
- Two-electron reduction leads to a cage opening, analogous to the bond-breaking found in adding electrons to an electron-precise cluster:
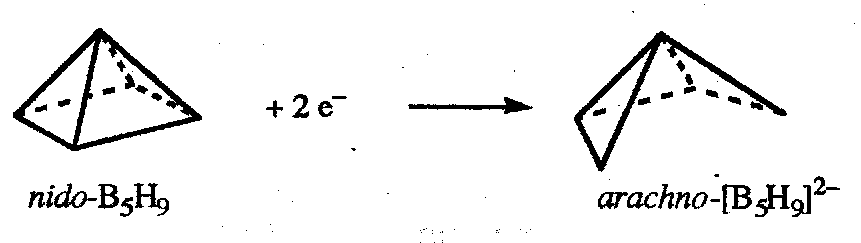
Some Borane Chemistry
Synthesis – mostly based on controlled thermolysis on B2H6. Mechanistically obscure and often very unselective, so separation of products required.
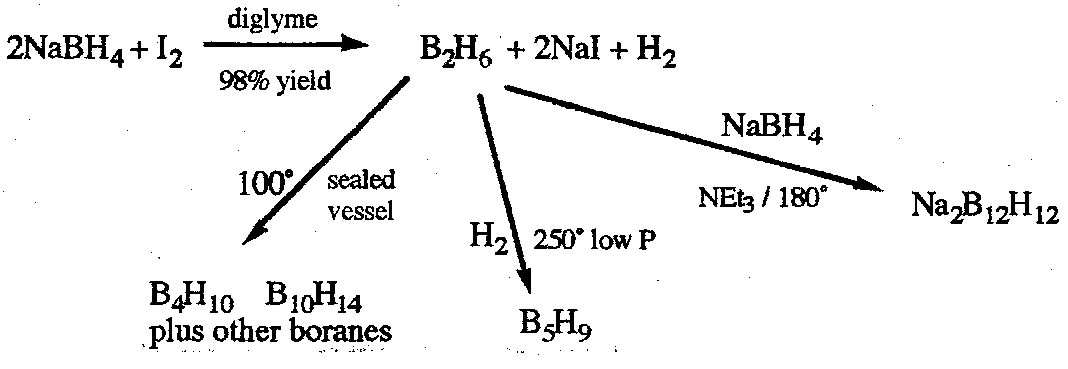
Characterisation – mass spec, IR, XRD, NMR all useful.
Properties:
Neutral boranes – highly inflammable (often spontaneously) and toxic. Reactivity generally arachno > nideo and decreases with size.
Weak Bronsted acidity, arachno > nido and increases with size.
React with Lewis Bases. Smaller boranes often cleave, as with B2H6.
There is a notable affinity for “soft” bases, L = Co, R2, R3P etc. With larger boranes substitution can occur.
Electrophilic substitution is possible, e.g. reaction with Cl2 or RCl in the presence of AlCl3 replaces one of more terminal H atoms with Cl or R respectively. In B5H9 calculations suggest that the apical boron is more negative than the others, and is the favoured site of substitution, although products slowly isomerise to ones with basal substituents, which are apparently thermodynamically more stable.
Closo-anions are kinetically much more stable, especially [B6H6]2-, [B10H10]2- and [B12H12]2-. Electrophilic substitution reactions are possible.
Solvent Properties
Water is known as the typical solvent, and is referred to as aqueous solution. There are however a number of other non-aqueous solvents that are often used. This is commonly when water cannot be used, either because it does not dissolve the substance, does not allow for certain reactions to take place, or simply reacts with the substances required.
The topic of solvents often merges with Acid-Base Chemistry, and as a consequence, Redox Chemistry. When analysing solvents it is important to consider two main properties:
- Melting Point and Boiling Point. This is obvious really, and these need to be both useful values around room temperature, as well as provide a long liquid range for use in chemical operations.
- Permittivity. This is typically referred to as the dielectric constant, ε. A high value is needed for ionic substance to dissolve readily. Water has a high value of 81.7, but there are higher.
The Dielectric Constant allows solvents to be large classified as highly polar, moderately polar, or non-polar.
Highly polar solvents dissolve ionic or polar substances. This is due to the Born Equation:
(This neglects donor-acceptor interactions).
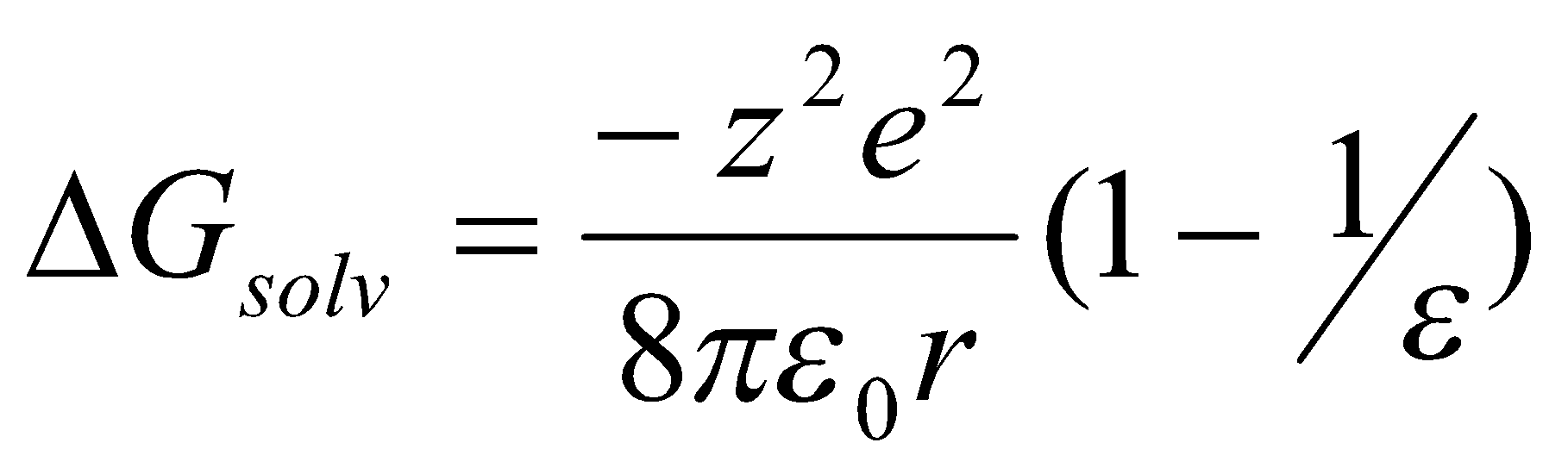
Moderately polar solvents for ion pairs (electrostatic attraction between two ions). Polarisability also becomes an important factor as polarity decreases, as strong dispersion forces also promote solubility, which is why CCl4 is a good solvent.
Donor/Acceptor interactions simply refers to the ability of a solvent to either donate lone pairs as a Lewis Base like NH3, which contributes to solvation of cations, or instead solvate anions by accepting electrons. This also encompasses H-bonding solvation, and solvolysis itself as an extreme case of interaction, where nucleophilic substitution occurs in donor solvents, e.g. OPCl3 + HX → X3PO + 3H+ + 3Cl-.
It should be noted that other transfers may occur, namely ion, halide or oxide transfer. Ion transfer principally refers to H+, and is named protic behaviour (although it is really autoionisation as an equilibrium of Bronsted Acidity/Basicity, e.g. 2H2O ⇌ H3O+ + -OH).
Halide transfer is a similar case, except X- (usually F) is the transferred ion and providing X makes the substance a base, while accepting it makes it an acid (Lewis definition). An example is 2BrF3 ⇌ BrF2+ + BrF4-, and this behaves as a strong fluorinating agent.
Oxide transfer is a similar case, except usually is studied in silicates. Oxides can be labelled as basic or acidic based on the same criteria as for halide transfer, such that e.g. CaO is basic while P4O10 is acidic. This is the Lux-Flood definition.
Redox Chemistry of Non-aqueous solvents also requires a mention. One of the significant uses of non-aqueous solvents is to extend the range of redox chemistry that can be carried out, overcoming other solvent limitations. For example, S42+ cannot be made in water, but can in other solvents. One such manifestation is the change in kinetics of the reactions, e.g. liquid ammonia runs slow redox reactions that can be studied more easily. Thermodynamically also, the variation from that in aqueous solution is due to the change in solvation energy, and also variation in the oxidation-reduction potentials of the solvent. Hence the effect on the electrode potentials varies depending on the solvent, e.g. Li (s) is too reactive in water or ammonia, but SO2 behaves as a solvent and an oxidant, and this is used to good effect in Lithium Batteries. For highly oxidising conditions superacids, sulphuric acid or SO2 should be used.
Considering now Ammonia as a non-aqueous solvent, it is a typical basic solvent which finds a wide variety of uses. It is physically similar to water but has a much smaller dielectric constant of 26.7. This means highly charged ions like carbonates tend to be insoluble, but other ionic compounds such as those of iodide will dissolve. Solubility may in fact increase due to stabilising interactions between the solute and the ammonia molecules, so it is not just a permittivity factor. These may arise e.g. from Metal + Ligand interactions (where the ligand is ammonia) or from a polarisable solute. Consequently ammonia can dissolve non-polar molecules more readily than water.
Precipitation will still occur in ammonia as in water. However, due to the changes in solubility, the precipitates may differ. For example, AgCl will dissolve in ammonia while KCl will precipitate out, whereas the opposite is true in water.
Another similar property is autoionisation: 2NH3 ⇌ NH4+ + NH2-. This allows for neutralisation reactions like water (H3O+ + HO-). This also obviously leads on to acid-base behaviour, and NH3 is also amphoteric, reacting exactly as water would in complex formation, e.g.
Zn2+ + 2OH- → Zn(OH)2 → Zn(OH)42- in excess –OH.
Zn2+ + 2NH2- → Zn(NH2)2 → Zn(NH2)42- in excess – NH2.
Ammonia is known as basic due to the properties of other acids when dissolved in it. Strong acids in water will react completely with ammonia, as will some weak acids (indicating they are stronger in ammonia). Similarly, non-acids in water often some weak acidic behaviour in ammonia. Likewise, bases in water are either insoluble or weak bases in ammonia, unless they are stronger bases than ammonia.
Solvolysis is also well-known ammonia and parallels the reactions of water. There is also a pH scale, where pH 0 is for 1M NH4+, which changes to pH 13 when [NH4+] = [NH2-] and pH 26 is neutrality (1M NH2-). Thus the principle differences between water and ammonia are in ammonia’s basicity, and its lower dielectric constant.
An interesting application of ammonia as a solvent is in dissolving alkali metals. A small amount of alkali metal produces a deep blue solution, regardless of the metal, while a larger amount forms a bronze metallic phase that floats on the blue, and shows conductivity and magnetism typical of a metal. Ammonia will evaporate from this to leave the alkali metal unchanged.
The blue solution has a density similar to NH3 alone, but shows paramagnetic properties and a g-factor close to the value for a free electron. It has been theorised that this corresponds to:
M + NH3 → M+ + [e(NH3)x]-.
This makes it an excellent one electron reducing agent and a strong base.
Typical acidic solvents are anhydrous H2SO4 or HF. Sulphuric Acid has a permittivity higher than water (110), while that of HF is roughly the same.
Sulphuric Acid shows extensive autoionisation, and dissolves ionic substances very readily. It also has a high viscosity (25 times that of water) which makes it practically very difficult to use to dissolve or crystallise compounds. HF does not have this problem.
HF has properties similar to water, but is highly reactive (etches glass!) and toxic. It autoionises as 3HF ⇌ H2F+ + HF2-, and the FHF- unit is good for dissolving fluorides. HF’s main advantage is that it is non-reducing and has a long liquid range, making it good for anodic oxidation reactions. This chiefly allows for the synthesis of fully fluorinated hydrogen derivatives.
As a rule most species will be basic sulphuric acid, including water. Many strong acids in water are bases in sulphuric acid, such as nitric acid. One of the few acids in a Sulphuric Acid solvent is HClO4, and also H2S2O7 (disulphuric acid). The only exceptionally strong acid is HB(HSO4)4.
It should be mentioned here that a mixture of HF and SO3, or the more facile SbF5 + 2HSO3F yield superacid, H2SO3F+. There are in fact a whole range of superacids, and they are typically very strong Bronsted Acids like Disulphuric Acid and very strong Lewis Acids such as SbF5, or a mixture of the two. The strongest of them all is H2F+, formed from HF + SbF5. Many superacids can protonate non-basic species like Xe, H2, CO2, Cl2 (but not O2, N2 or Ar). They also cleave organic compounds such as candle wax, which forms the t-butyl cation.
As a summary of protic solvents, it is noted that they show a great similarity to acid-base behaviour. This is obviously typical of the Bronsted definition due to the presence of ionisable H’s. Factors that affect them are thus: 1) inherent acid/base of solvent, 2) inherent acid/base of solute, and 3) solute-solvent interaction (equilibrium = weak, complete = strong).
Aprotic solvents, on the other hand, do not have transferable Hydrogen. They can be typically classified into three groups:
|
Non-polar, non-solvating, don’t autoionise |
Polar, don’t autoionise |
Highly polar, autoionise |
|
e.g. hexane, CCl4 |
e.g. MeCN, DMSO, SO2 |
e.g. BF3, OPCl3 |
|
Solvent plays the minimum role in the chemistry |
Polarity confers good coordination chemistry (most are basic). |
Often very reactive themselves. |
The middle group of polar, non-ionising solvents, bridge the gap between the two extremes. They are typically classified based on their Donor Number, an arbitrary system for accounting for their action as ligands (so their basicity/polarity). It does not account for Hard/Soft interactions though, so is limited. It is defined as the –ΔH of the reaction B + SbCl5 → BSbCl5.
One of the more important aprotic solvents is sulphur dioxide. It dissolves molecular liquids or solids more readily than ionic substances. This is due to a low dielectric constant, which is shown by ion-pairing and poor conductance of ionic solutes. Its main advantage is a long liquid range with low viscosity, so is a useful preparative solvent. It is not particularly acidic or basic either, so goof for acid-base reaction reagents.
Donor / Acceptor Chemistry
Definitions:
- A donor (or Lewis Base) is a species with an electron pair available for sharing.
- The electrons involved can sometimes come from bonding orbitals, e.g. benzene and BH4- donations.
- An acceptor (or Lewis Acid) is a species with a vacant orbital and coordination site available for accepting the electron pair from a donor.
- Sometimes the empty orbital involved may be an antibonding one of the acceptor, e.g. σ* of I2 is relatively low-lying and gives rise to acceptor properties, e.g. I3- formation.
- H-bonding can also be regarded as a donor/acceptor interaction.
Modes of Interaction
Electrostatic Forces – anion-cation, ion-dipole, and dipole-dipole interactions can be important, e.g. in complexes between metal cations and anions or polar ligands such as H2O.
Dispersion Forces – may be significant between large polarisable species.
Frontier-Orbital Overlap – involves the HOMO of the donor and the LUMO of the acceptor. Note that essentially a polar covalent bond is formed, except that in a donor/acceptor complex both electrons are regarded as coming from the donor, rather that one from each fragment.
Formation of some complexes (e.g. transition metal carbonyls) involves a two-way donor/acceptor interaction, with the ligand acting as a σ-donor through its lone pair, and a π-acceptor through empty π* orbitals.
Consequences
- Solvation of ions and polar molecules (see above).
- Complexes of metal ions and atoms.
- Complexes between neutral molecules (adducts).
- Complex anions and cations formed by ion transfer.
- Self-association of molecules can occur when they have both donor and acceptor parts. This includes H-bonding in liquids and formation of dimers or other oligomers with halogen bridges.
- Nucleophilic substitution can be consequence of initial donor/acceptor complex formation.
- Bronsted Acidity of donors with ionisable H+ is increased on complex formation, e.g. BF3.2H2O undergoes autoprotolysis on melting. Also, superacids (see earlier), and acidity of metal cations in water.
- Perturbation of vis/UV absorption spectrum arises from shift in orbital energies on complex formation, and the possibility of charge-transfer transitions from donor to acceptor based MOs. This has been particularly studied with I2 complexes.
Donor and Acceptor Strengths
The relative strengths of different donors or acceptors depend to some extent on the partner.
Acceptor Strengths towards N, O and F donor generally follow the order:
BF3 < BCl3 < BBr3 < Bi3.
With most non-metals the reverse order of halides is found:
BF3 > SiF4 < PF5 < AsF5 < SbF5, and PX3 < PX5.
Donors strengths towards H+ and halide acceptors generally follow the orders:
R3N > R3P > R3As
F- > Cl- > Br- > I-
R3N > R2O
R3C- > R2N- > RO- > F-
The Hard and Soft Acid and Base (HSAB) principle is an attempt to systematise some of the different preferences observed. See Coordination Chemistry Notes for details on this.
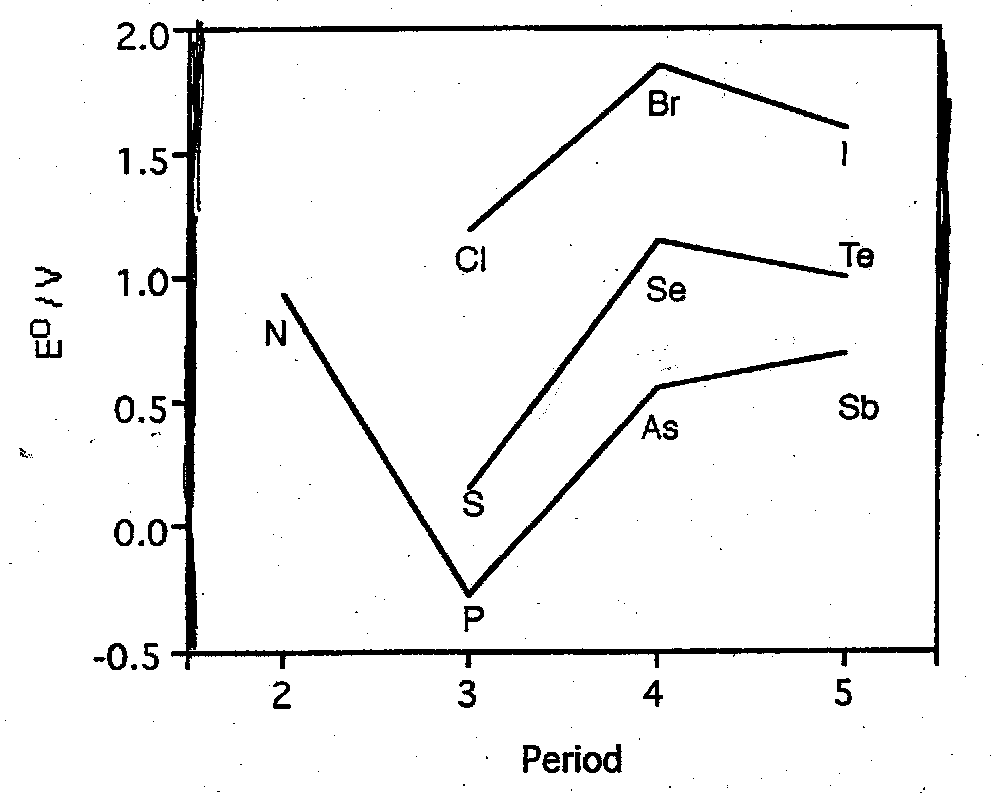
High Oxidation States
Some Group Trends
Redox Potentials N/(N-2) where N is ground valence:



Structures of oxo-species in maximum oxidation state:
Period 2 : HNO3 (strong acid), NO2+, NO43- (only in solid salts).
Period 3: H3PO4, H2SO4, HClO4.
Period 4: H3AsO4, H2SeO4, HBrO4
Period 5: [Sb(OH)6]-, Te(OH)6, IO(OH)5 ⇌ IO4-.
Recall Pauling’s Rules for oxoacid strengths in water.
“Alternation Effect” resulting from periodic table given irregular IE trends.
Br(VII) is more strongly oxidising than either Cl(VII) or I(VII).
Stability of Group 15 Pentahalides:
N(V): no NX5 molecules, but NF3 + F2 + SbF5 → [NF4]+[SbF6]-.
P(V): PX5 with X = F,Cl,Br; but only [PI4]+I- solid (molecule not stable).
As(V): only AsF5 (v. strongly oxidising known under normal conditions. AsCl5 decomposes above -50oC.
Sb(V): SbF5 and SbCl5 known.
Cationic Chemistry of non-metals
Oxocations (NO+, NO2+) and many cationic halide complexes are known. Monatomic cations formed by non-metals are extremely strong acceptors and are known only complexed with strong donors Formation of uncomplexed polycations requires:
- very strong oxidising agents, e.g. AsF5 or S2O6F.
- counterions with only very weak donor properties, e.g. [AsF6]-.
- Solvents which provide or tolerate these conditions, e.g. superacids or liquids SO2.
Examples of Reactions are:
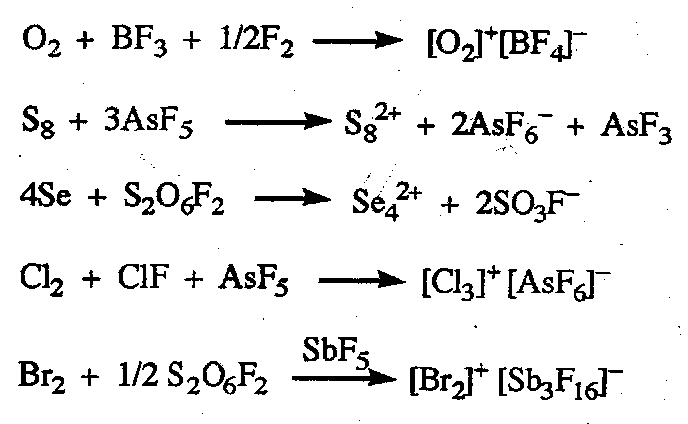
Xenon Chemistry
Starting point is the direct reaction with F2:
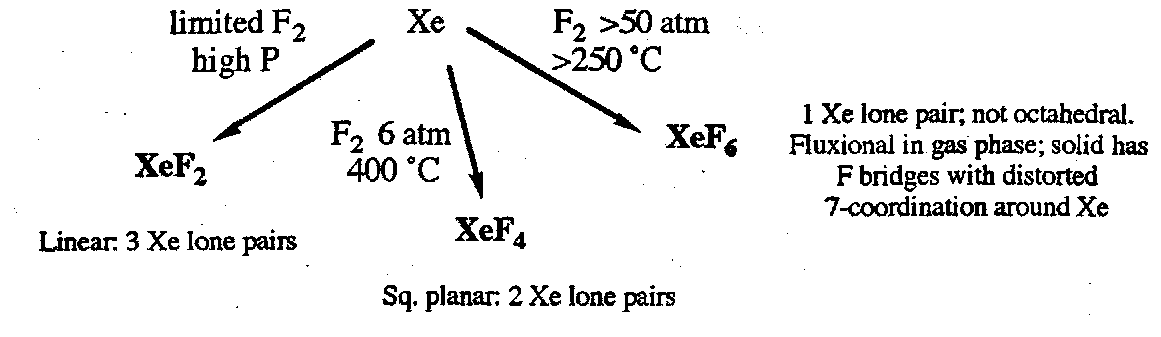
Xenon fluorides are strong fluorinating agents, are violently hydrolysed, and react with silica. Reactions with F- donors or acceptors give anionic or cationic fluoride complexes respectively. Examples:
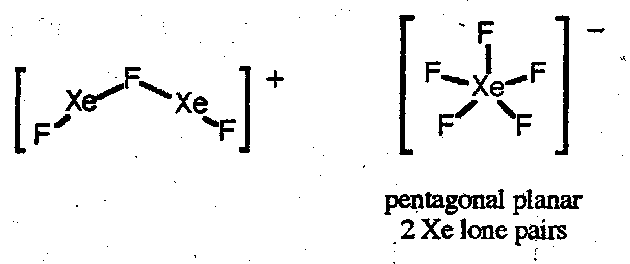
Reactions with SbF5 also gives the dixenon cation [Xe2]+:
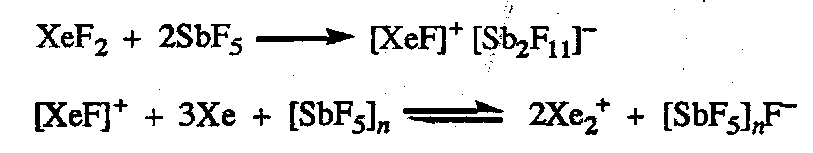
Green paramagnetic Xe2+ characterised by UV, Raman and EPR spectroscopy. Xe-Xe distance of 308pm.
Hydrolysis reactions giving xenon oxygen compounds and oxofluorides are shown below. All oxides are thermodynamically unstable, and XeO3 is dangerously explosive.
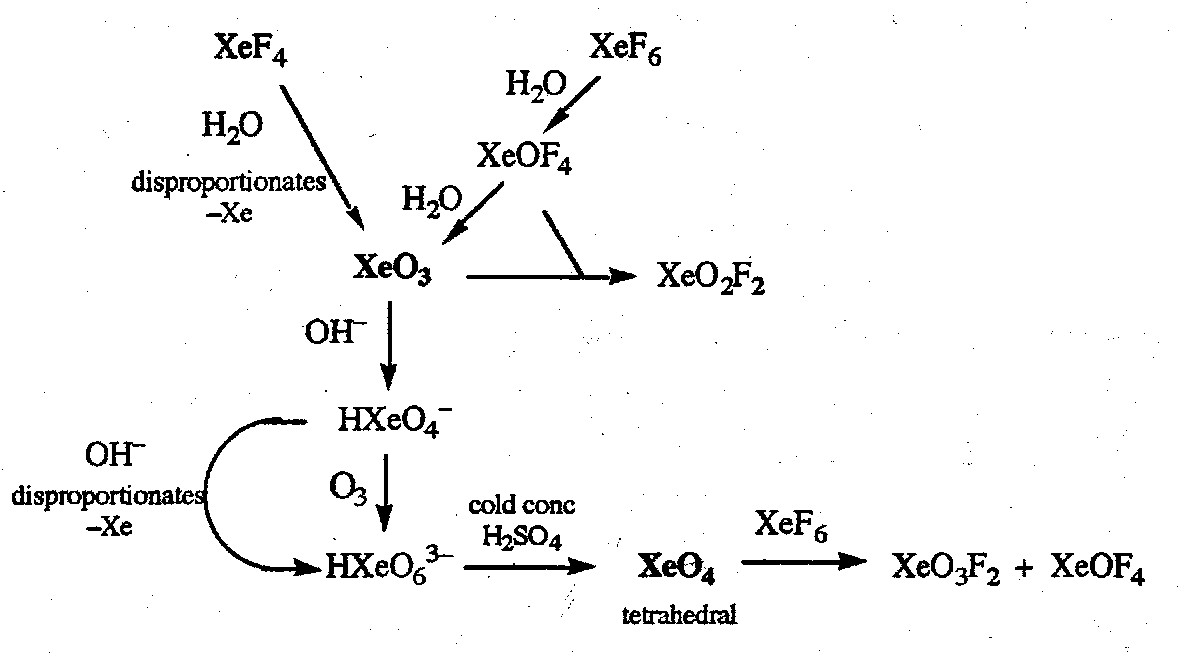
To stabilise Xe compounds, need electron withdrawing groups around it.
Compounds with Xe-N and Xe-C bonds can be prepared. Examples are:
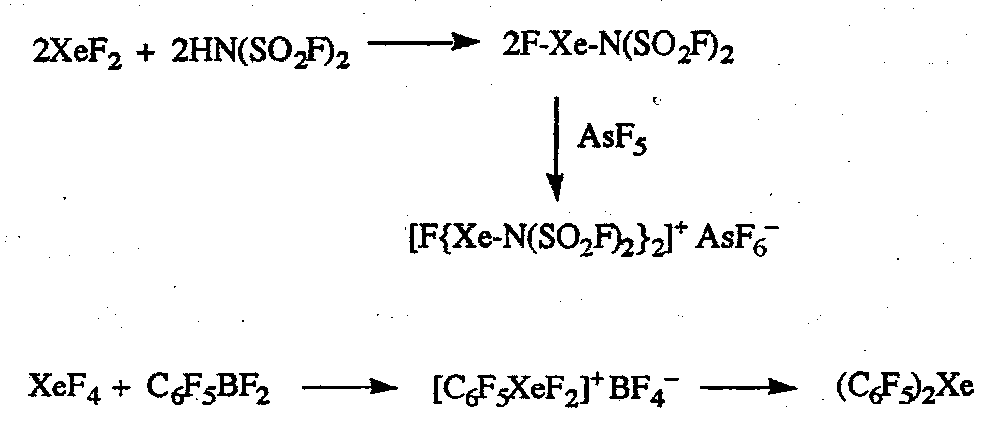
Also recently prepared compound is the remarkable Au(II) complex [AuXe4]2+. Xe is a weak donor, so need a very soft cation.
Conventional compounds of krypton are limited to very unstable KrF2 and some [KrF]+ compounds. The lesser stability compared with Xe parallels the higher IE of Kr, as expected from 3-centre bonding models which require positive charge on Xe or Kr. However, ions such as ArH+ and KrH+ are well-known in the gas phase, and compounds such as [ArH]+F- have recently been prepared by low temperature matrix isolation. They are only kinetically stable, decomposition to Ar + HF being thermodynamically very favourable.
A Comparison of N and P
Three major features:
- the radius of phosphorus is 50% greater than that of nitrogen
- the ionization energies decrease from N to P
- the bond energy trends are different
The oxidation states for both elements vary from +5 to -3, but the stability trends are different.
Valence shell expansion: the coordination number of nitrogen is ≤ 4, but phosphorus is larger and the C.N. can be up to 6: PF6- and PCl6- but NH4+ and NF4+.
Bond enthalpies:
N-H vs. P-H chemistry
The N-H bond is stronger than the P-H bond (386 vs. 321 kJ mol-1), so despite a lower ΔHoatm for P, PH3 is thermodynamically unstable. PH3 is pyrophoric (kinetics). NH3 also a stronger base.
Lone-pair repulsion
Nitrogen:
Weakens N-X bonds in many nitrogen compounds resulting in thermodynamic instability,
e.g., the oxides and halides.
Phosphorus:
Much reduced in P-X bonds (also dπ-pπ bonding, see below), hence stronger bonds and thermodynamically stable halides and oxides.
Compare:
Hydrolysis of NCl3 gives NH3 and Cl2O, whereas PCl3 gives H3PO3 and HCl.
pπ-pπ bonding
Nitrogen:
Very important feature of nitrogen chemistry. Coupled with weak N-X single bonds, nitrogen compounds tend to be monomeric or dimeric, e.g., elemental nitrogen (N2), oxides, nitrates and nitrites.
Phosphorus:
Quite strong multiple bonds but single bonds even more important.
Hence many phosphorus compounds "polymerize", e.g., the oxides (N2O3 and N2O5 vs. P4O6 and P4O10), oxy-anions, suphides [note: NO is monomeric but N4S4 and P4S3, P4S4 etc], elemental phosphorus (P4), and nitrogen compounds [e.g., (NPCl2)4].
dπ-pπ bonding
Open only to phosphorus, and particularly in compounds with the more electronegative elements N, O, F.
Strengthens both P-X and P=X bonds and hence chain or cyclic structures for the oxides and oxy-anion salts.
Kinetic effects too:
X3N-O X3P=O
Normal N-O bond length
high polarity
low energy
high reactivity
Short P=O bond
low polarity
high energy
inert.
Oxidation states
- Negative oxidation state stability N > P
- Positive oxidation states P > N
- Oxidation states: NCl3 very unstable but PCl3 stable; N2O5, NO3, NF4 but P4O10, PF5, PCl5, PBr5; nitric acid is very oxidizing but phosphoric acid is not.
- In aqueous solution: refer to oxidation state diagram.
Note: H3PO2 is in fact H2P(=O)OH, and phosphorous acid is not P(OH)3 but HP(=O)(OH)2 - an indication of the P=O bond strength.
- Donor properties of NR3 and PR3 in complex formation.
Comparison of Oxygen with Sulphur
Trends are similar to those in N/P comparison.
Decrease in Zeff from O to S
- no positive oxidation states for O (except O2+ in solid state though not in solution) but sulphur from -2 to +6 and forms Sn2+ (n = 4, 8, 19) in strong acids
- oxides more ionic than sulphides
- less hydrogen bonding in S-H compared to O-H
- acidity of H2O
vs H2S (but note HF vs HCl)
Oxygen is much smaller than sulphur.
At short distances for covalent bond formation, the coordination number of oxygen rarely exceeds 2 (H3O+ but no F3O+, compare with NF4+). The coordination number of sulphur can be up to 6, e.g., SF6, SF5Cl.
Note: SF6 is kinetically
stabilized to hydrolysis.
Catenation
lone-pair repulsion weakens O-O single bonds, catenated oxygen compounds very unstable. The S-S single bond is quite strong (266 kJ mol-1) => increased catenation on going to sulphur.
- O2(g) vs. S8(s)
- H2O2 is unstable and strongly oxidizing but H2Sn with n up to 100 known; O-O bonds used in biological oxidations but S-S bonds stabilize protein folding.
- ozonides O3- very unstable but many polysulphides SnX.
- S-S bonds in dithionite and polythionites.
Bond Strengths
The strength of the S-S bond is comparable to that of the S-Cl bond (271 kJ mol-1) and greater than both of S-Br and S-I. Hence S2Cl2 but not SCl2, and even S2I2 is unstable.
Note: OCl2 but not
O2Cl2.
Compounds with nitrogen:
Molecular NxOy but (SN)z due
to π-bonding.
dπ→pπ bonding
In S-F, S-O and S-N bonds.
Note: SO unstable with respect to S8 and SO2; SF2 with respect to S8 and SF4.
I am happy for them to be reproduced, but please include credit to this website if you're putting them somewhere public please!