Lanthanides and Actinides
Mostly Descriptive Chemistry, but discusses why the chemistry occurs as well. There is some more detail on Uranium at the end. Adapted from Dr Heyes' Lecture Notes
Lanthanides & Actinides Notes
General Background
Mnemonics
Lanthanides
|
Lanthanide |
Chemistry |
Presents |
No |
Problems |
Since |
Everyone |
Goes |
To |
Doctor |
Heyes' |
Excruciatingly |
Thorough |
Yearly |
Lectures |
|
La |
Ce |
Pr |
Nd |
Pm |
Sm |
Eu |
Gd |
Tb |
Dy |
Ho |
Er |
Tm |
Yb |
Lu |
Actinides
|
Although |
Theorists |
Prefer |
Unusual |
New |
Proofs |
Able |
Chemists |
Believe |
Careful |
Experiments |
Find |
More |
New |
Laws |
|
Ac |
Th |
Pa |
U |
Np |
Pu |
Am |
Cm |
Bk |
Cf |
Es |
Fm |
Md |
No |
Lr |
Principal Characteristics of the Rare Earth Elements
- Occur together in nature, in minerals, e.g. monazite (a mixed rare earth phosphate).
- Very similar chemical properties. Found combined with non-metals largely in the 3+ oxidation state, with little tendency to variable valence.
- Small difference in solubility / complex formation etc. of M3+ are due to size effects.
Traversing the series r(M3+) steadily decreases – the lanthanide contraction.
Difficult to separate and differentiate, e.g. in 1911 James performed 15000 recrystallisations to get pure Tm(BrO3)3!
f-Orbitals
The Effective Electron Potential:
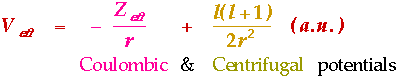
- Large angular momentum for an f-orbital (l = 3).
- Large centrifugal potential tends to keep the electron away from the nucleus.
- Aufbau order.
- Increased Z increases Coulombic attraction to a larger extent for smaller n due to a proportionately greater change in Zeff.
- Reasserts Hydrogenic order.
This can be viewed empirically as due to differing penetration effects.

Radial Wavefunctions Pn,l2 for 4f, 5d, 6s in Ce
4f orbitals (and the atoms in general) steadily contract across the lanthanide series.
Effective electron potential for the excited states of Ba {[Xe] 6s 4f} & La {[Xe] 6s 5d 4f} show a sudden change in the broadness & depth of the 4f "inner well".
For Ba (Z = 56) 4f is an outer orbital with 4f close to its value for the H atom.
For La (Z = 57) 4f is an inner orbital with 4f ca. 0.7ao.
Though only for the next atom, Ce (Z = 58) is the 4f electron of sufficiently high binding energy to appear in the ground state configuration.
Which Elements are d-block or f-block?
Most current Periodic tables have:-
- La as first 5d transition element
- Ac as first 6d transition element
Reasons?
Possibly erroneous (early) interpretation of atomic spectra – misleading electronic configurations?
Some ground state electronic configurations:
|
Calcium [Ar]4s2 |
Scandium [Ar]4s23d1 |
|
Strontium [Kr]5s2 |
Yttrium [Kr]5s24d1 |
|
Barium [Xe]6s2 |
Lanthanum [Xe]6s25d1 |
|
Ytterbium [Xe]6s24f14 |
Lutetium [Xe]6s24f145d1 |
|
Radium [Rn]7s2 |
Actinium [Rn]7s26d1 |
|
Nobelium [Rn]7s25f14 |
Lawrencium [Rn]7s25f146d1 |
In each case: differentiation by (n-1)d1 - as expected for the start of a transition series.
Lutetium and Lawrencium are just as good candidates to be the first elements of the 3rd and 4th transition series as Lanthanum and Actinium.
Some suggestions why Lu might best regarded as the first 5d transition element.
- Periodic Trends in Various Properties.
- Structures of Metal, Metal Sesquioxide (M2O3) and Metal Chloride (MCl3).
- Similarities for Sc, Y, Lu.
- Differences from La.
Revised Medium-Block Format Periodic Table
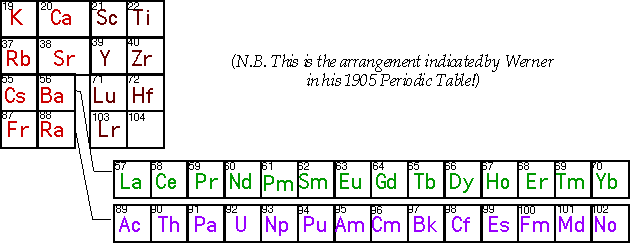
The Lanthanides
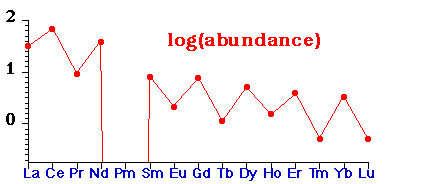
Abundance & Distribution
- Not especially Rare(!), except Promethium which is produced artificially, e.g. La, Ce & Nd are more common than Pb.
- Most-common minerals:
monazite (mixed La, Th, Ln phosphates) – widely-distributed, concentrated in sand & river beds due to relative insolubility.
bastnaesite (a La, Ln fluorocarbonate MIIICO3F) – a vast deposit in Sierra Nevada, USA.
- Abundance of lanthanides in nature. Shows even-odd alternation with atomic number mirrored by several/few alternation of number of stable isotopes with even/odd Z.
Extraction
The extraction of the Lanthanides from minerals by:
- Alkali Digestion of Monazite/Xenotime.
Monazite and Xenotime can also be opened-out through an Acid Route
- Acid Dissolution of Bastnaesite
Separation
2/3 of world production is actually used mixed in the proportions occurring naturally in the ore.
1. Cerium & Europium may be extracted Chemically:
Oxidise only Ce to M4+ by HClO or KMnO4, then precipitate as CeO2 or Ce(IO3)4.
On action of Zn/Hg only Eu forms a stable M2+ that doesn't reduce H2O, then isolate by precipitation as EuSO4.
2. Separation by Fractionation:
Small Scale methods used originally:
- Fractional Crystallization of e.g. Ln(NO3)3.2NH4NO3.4H2O or Ln(BrO3)3
- Fractional Thermal Decomposition of e.g. Ln(NO3)3
Current Small Scale Lab Separation:
- Ion-Exchange Displacement Column
Ln3+(aq) are strongly adsorbed by a cation-exchange resin, add a ligand, typically chelating, e.g. EDTA.
Ligand binds most strongly to smallest ion, e.g. the binding constants of the Ln(EDTA) complexes (right).
Current Large Scale Industrial Separation:
Solvent Extraction
Ln3+(aq) is extracted in a continuous counter-current process into a non-polar organic liquid (e.g. kerosene).
Solubility of Ln3+ in organic solvent increases with its relative atomic mass.
The Metals
Production of Elemental Metals
La, Ce, Pr, Nd, Gd:
2MCl3 + 3Ca → 2M + 3CaCl2 (T > 1000°C)
Tb, Dy, Ho, Er, Tm, Y:
2MF3 + 3Ca → 2M + 3CaF2 (MCl3 is too volatile)
Pm:
PmF3 + 3 Li → Pm + 3LiF
Eu, Sm, Yb:
M2O3 + 2La → 2M + La2O3 (MCl3 reduced to MCl2 by Ca)
Mischmetall (mixed light Ln) :
electrolysis of fused LnCl3/NaCl with graphite anode & graphite or steel cathode.
Structures of Elemental Metals

Properties of the Metals
- Silvery white, but tarnish in air.
- Rather soft (later M are harder).
- High mp & bp.
- Very reactive: (I1 + I2 + I3) comparatively low.
- Burn easily in air, but slowly in cold. Burn at T > 150 °C.
- Exothermic reaction with H2 → MHn (n = 2,3, often results in defect states).
- React readily with C, N2, Si, P, halogens & other non-metals.
- Form binaries on heating with most non-metals (e.g. LnN, Ln2S3, LnB6, LnC2, ...).
Uses of Metals
<1% Mischmetall or Ln silicides improves strength & workability of low alloy steels for plate and pipes (also used in Mg alloys).
Mischmetall (50% Ce, 25% La, 25% other light lanthanides) is pyrophoric - alloyed with 30% Fe it is used in lighter flints.
Oxidation State Preferences
Chemistry is principally of Ln3+.
Examine Thermodynamic Parameters: Ionisation Energies, Heats of Hydration and Atomisation.
Ionization
For any given Lanthanide:
- As successive electrons are removed from neutral Ln the stabilizing effect on the orbitals is related to their principal quantum number, 4f > 5d > 6s.
- For Ln2+ (except for La & Gd) the configuration is [Xe]4fn
- For Ln3+ the configuration is always [Xe]4fn
- The 4f binding energy is so great that remaining 4f electrons are regarded as "core-like" (i.e. incapable of modification by chemical means), except Ce.
Therefore in almost all cases Ln3+ provides the best energetics:
Observing trends across the Lanthanide Series:
- The general trend is for increasing ionization energies with increasing Z (i.e. with increase in Zeff)
- Marked Half-Shell Effects - magnitude as n in In
- Also Quarter/Three-Quarter Shell Effects (compare with transition metals - these are not seen clearly with dn configurations).
Explanation:
Inter-electronic repulsion is related not just to electron pairing but also to angular momentum of the electrons.
e.g. in Pr2+ (4f3) → Pr3+ (4f2) ionization removes repulsion between e- of like rotation, whereas Pm2+ (4f4) → Pm3+ (4f3) removes the stronger repulsion between e- of unlike rotation (latter Ionization Energy is correspondingly lower - hence the local minimum in the I3 graph at Pm).
The three-quarter effect is the bigger: interelectronic repulsion is bigger in smaller Lnn+
Atomization
ΔatmH follows the inverse trend to I3, and therefore also to (I1 + I2 + I3).
Metallic bonding is correlated with ease of ionization to Ln3+ state.
This trend is modified slightly due to the different structures of the Ln metals.
Some Thermodynamic Observations (Ionic Model style)
The trends in the formation of LnIII Compounds, ΔfH for LnX3(s) or Ln3+(aq) [ E°(Ln3+(aq) / Ln(s)) ] depends on the balance between:
Energy Supplied to effect:
Ln(s) → Ln(g) → Ln3+(g) + 3e- [ ΔatmH + I1 + I2 + I3 ]
vs.
Energy gained from:
Ln3+(g) + 3X-(g) → LnX3(s) [ ΔLH(LnX3(s)) ]
or Ln3+(g) → Ln3+(aq) [ΔhydH(Ln3+)]
The energies determining trends in E°(Ln3+(aq)/Ln(s)) are graphed below:
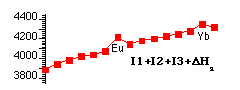
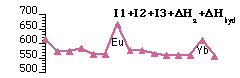
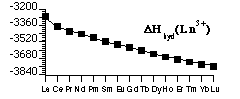
Production of Ln3+(g) shows:
- A smooth trend based on size effects (trend based on Zeff).
- Shell structure effects superimposed with clear maxima at half-shell (f7) and full-shell (f14).
- Also smaller quarter and three-quarter shell effects.
Hydration Energy of Ln3+ (also Lattice Energies of LnX3(s)) shows:
- Only a smooth ionic-size-based trend (the trend based on Zeff) and no shell structure effects.
- Balance of trends in Ionization + Atomization Energies with Hydration (Lattice) Energy removes size effects.
- Leaves only the Shell effects - see values of ΔfH(Ln3+(aq)).
Overall:
The most important energy correlations are with I3
Exceptions to +3 rule can also be rationalized.
Occurrence of +4 oxidation state
Predicted from [ΔatmH + I1 + I2 + I3 + I4] which follows trends in I4.
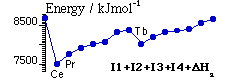
Ce, Pr → Ce4+ [4f0], Pr4+ [4f1] - early in series 4f orbitals still comparatively high in energy.
Tb → Tb4+ [4f7 valence shell] - half shell effect.
Occurrence of +2 oxidation state
Predicted from [ΔatmH + I1 + I2] which follows trends in ΔatmH, which is reverse of trend in I3.
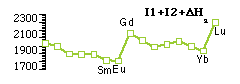
Eu, Sm, Yb → Eu2+ [4f7], Sm2+ [4f6], Yb2+ [4f14] - clear influences of electronic shell structure & from ΔatmH.
General Features of Lanthanide Chemistry
1. similarity in properties, with gradual changes occurring across the lanthanide series: a size effect from the Lanthanide Contraction
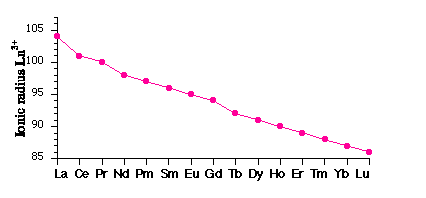
Causes:
Poor screening of nuclear charge by 4f electrons → steady increase in Zeff.
Relativistic effects influence the shielding characteristics of inner electrons.
2. Primarily the 3+ oxidation state adopted for all elements
Redox chemistry is commonly encountered only for Eu (3+/2+) and Ce (4+/3+).
Some solids formulated as LnII compounds actually contain Ln3+ & delocalized e-.
3. Coordination chemistry is not especially extensive
Chelating ligands are preferred.
4. Bonding on coordination is primarily ionic in character
Complexes undergo rapid ligand exchange.
Why is the bonding so ionic?
4fn electrons are contracted into the core and unable to participate in bonding.
Other implications from lack of covalent bond-forming orbital-availability:
- No π-backbonding occurs.
- No simple carbonyl species (except in Ar matrix at 10 K).
- Cyclopentadienyls are ionic in nature [c.f. Ln(C5H5)3 vs. Fe(C5H5)2].
- Lanthanide organometallics have different properties from transition metal equivalents.
5. Ln3+ cations display typical a-class (hard) properties
preference for O-donor ligands.
Why not N too? O-donor ligands are more likely to be charged (importance of ionic bonding to lanthanides!)
6. Binding to water is common
Such that H2O is often found included in products isolated from (aq).
7. Coordination numbers are high
> 6, typically 8, 9,... (up to 12 found).
8. Coordination polyhedra are often ill-defined
Determined by ligand requirements, not by bonding requirements.
No confirmed examples of isomerism.
Solid state structures of binaries are often rather different from those of other metals.
9. Ligand Field Effects are very small
Pale Colours from weak, narrow forbidden f-f optical transitions.
M3+ ions are:
Colourless (La, Ce, Gd, Yb, Lu)
Green (Pr, Tm)
Lilac (Nd, Er)
Yellow Pink (Pm, Ho)
Yellow (Sm, Dy)
Pale Pink (Eu, Tb)
Magnetic properties have spin-orbit coupled contributions (spin-orbit coupling >> ligand field splittings)
Magnetic & Optical properties are largely independent of environment (e.g. similar spectra in gas/solution/solid).
Renewed Technological interest in Lanthanides is mainly in optical/magnetic materials .
Solution Chemistry
Solubility
- Ln3+ are not especially soluble in water.
- No simple relationship of solubility to cation radius.
- Depends on small difference between large solvation and lattice energies.
- Depends on entropy effects.
Hydrated Lanthanide Ions
- Primary hydration numbers in (aq) are 8, 9.
- Primary hydration number decreases with Lanthanide Contraction.
- Secondary hydration number increases with Lanthanide Contraction.
- Increased polarization of primary hydration sphere by a smaller cation enhances hydrogen bonding to water in the secondary hydration sphere.
- Aqua ions hydrolyze - increasingly so from La to Lu as they become smaller.
Ln(H2O)n3+ + H2O → Ln(H2O)n-12+ + H3O+
- Salts with common anions frequently contain Ln(H2O)93+ with tri-capped trigonal prismatic geometry.
Coordination Compounds
Strongly complexing, chelating ligands necessary to yield isolable products from (aq).
Some O-donor chelating ligands that form complexes include NO3- , which binds in its chelate mode.
Notable for high Coordination Numbers.
Ce(NO3)52- - 10-coordinate bicapped dodecahedron.
Ce(NO3)63- - 12-coordinate icosahedron.
Other favoured ligands include: Oxalate, Citrate, Tartrate.
Classic bidentate complexes formed as Ln(L-L)3L' (L' = H2O, py, etc...) or Ln(L-L)4 dehydrate in vacuo → Ln(L-L)3 [ now coordinatively unsaturated ].
Ln(L-L)3 with bulky R are thermally stable, volatile & sublimable, and soluble in non-polar solvents.
- Widely used as NMR shift reagents. Polar molecules may coordinate Ln(L-L)3, their NMR resonances are perturbed by the paramagnetism of Ln, e.g. Eu(facam)3.
- "Anti-knock" activity as petroleum additives.
Solution Chemistry of Other Lanthanide Oxidation States
Ln(IV)
Cerium is the only Ln4+ with significant aqueous or coordination chemistry.
E° (Ce4+(aq) / Ce3+(aq)) = 1.72 V (others estimated at 2.9 V).
Prepared by the action of a strong oxidizing agent, e.g. S2O82-, on Ce3+(aq).
Widely used as an oxidant itself, e.g. quantitative analysis / organic chemistry.
E° (Ce4+/Ce3+) is markedly dependent on complexation and hydrolysis.
Strong oxidizing agent in perchloric acid solution. In other acids coordination occurs, e.g. Ce(NO3)62- is generally used for oxidations as its NH4+ salt
Change in pH:
Hydrolysis to Ce(OH)3+ occurs, then polymerization. Ultimately precipitation of yellow gelatinous CeO2.xH2O.
4+ charge stabilizes halogeno-complexes e.g. CeF84-.
Ln(II)
Significant solution chemistry of Ln2+ is essentially confined to SmII, EuII, YbII
Preparation:
electrolytic reduction of Ln3+(aq).
Eu2+ (the most stable LnII) is prepared by reduction of Ln3+(aq) with Zn/Hg.
Properties:
Ln2+ Aquo-ion colours:
Sm2+ blood-red
Eu2+ colourless
Yb2+ yellow
- Ln2+(aq) are readily oxidized by air, but Eu2+(aq) is easily handled.
- Sm2+(aq) & Yb2+(aq) reduce water.
- Eu2+(aq) is relatively stable in the dark.
- Carbonate and sulphate salts have been isolated.
- Sm2+ and Yb2+ salts are susceptible to oxidation by their water of crystallization.
- Eu & Yb dissolve in liquid ammonia to give intense blue, highly reducing solutions containing [Ln(NH3)x]2+ and solvated electrons. Solutions decompose on standing, precipitating the amide Ln(NH2)2
- Properties of Ln2+ are closely-related to those of the alkaline earths. In particular Eu2+ is often likened to Ba2+.
Similar Salt Solubilities (like Ba, sulphates are insoluble, hydroxides are soluble).
Behaviour in liquid NH3 is very similar.
Similar Coordination Chemistry (Not extensive / Hard ligands)
BUT very different redox chemistry!
Magnetism & Spectra
|
Ln |
Ln3+ configuration |
Ground State |
No. of unpaired e- |
Colour |
gJ√(J(J+1)) |
Observed μeff/μB |
|
La |
4f0 |
1S0 |
0 |
colourless |
0 |
0 |
|
Ce |
4f1 |
2F5/2 |
1 |
colourless |
2.54 |
2.3 - 2.5 |
|
Pr |
4f2 |
3H4 |
2 |
green |
3.58 |
3.4 - 3.6 |
|
Nd |
4f3 |
4I9/2 |
3 |
lilac |
3.62 |
3.5 - 3.6 |
|
Pm |
4f4 |
5I4 |
4 |
pink |
2.68 |
- |
|
Sm |
4f5 |
6H5/2 |
5 |
yellow |
0.85 |
1.4 - 1.7 |
|
Eu |
4f6 |
7F0 |
6 |
pale pink |
0 |
3.3 - 3.5 |
|
Gd |
4f7 |
8S7/2 |
7 |
colourless |
7.94 |
7.9 - 8.0 |
|
Tb |
4f8 |
7F6 |
6 |
pale pink |
9.72 |
9.5 - 9.8 |
|
Dy |
4f9 |
6H15/2 |
5 |
yellow |
10.65 |
10.4 - 10.6 |
|
Ho |
4f10 |
5I8 |
4 |
yellow |
10.6 |
10.4 - 10.7 |
|
Er |
4f11 |
4I15/2 |
3 |
rose-pink |
9.58 |
9.4 - 9.6 |
|
Tm |
4f12 |
3H6 |
2 |
pale green |
7.56 |
7.1 - 7.6 |
|
Yb |
4f13 |
2F7/2 |
1 |
colourless |
4.54 |
4.3 - 4.9 |
|
Lu |
4f14 |
1S0 |
0 |
colourless |
0 |
0 |
Magnetic Properties
- Paramagnetism.
- Magnetic properties have spin & orbit contributions (contrast "spin-only" of transition metals).
- Magnetic moments of Ln3+ ions are generally well-described from the coupling of spin and orbital angular momenta - Russell-Saunders Coupling Scheme.
- Spin orbit coupling constants are typically large (ca. 1000 cm-1).
- Ligand field effects are very small (ca. 100 cm-1).
- only ground J-state is populated.
- spin-orbit coupling >> ligand field splittings.
- magnetism is essentially independent of environment.
- Magnetic moment of a J-state is expressed by the Landé formula:
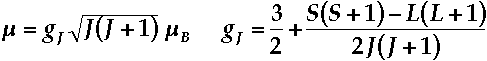
Ln3+ Magnetic Moments compared with Theory
Landé formula fits well with observed magnetic moments for all but SmIII and EuIII.
Moments of SmIII and EuIII are altered from the Landé expression by temperature-dependent population of low-lying excited J-state(s).
Uses of Ln3+ Magnetic Moments?
NMR Shift Reagents - paramagnetism of lanthanide ions is utilized to spread resonances in 1H NMR of organic molecules that coordinate to lanthanides.
Ferromagnetism / Anti-Ferromagnetism / Ferrimagnetism
Lanthanide metals and alloys have interesting ordered magnetism effects.
SmCo5, Nd2Fe14B permanent magnets - FERROMAGNETIC
- light weight.
- high saturation moments.
- high coercivity.
- high magnetocrystalline anisotropy.
- Superior performance magnets for magnetic bearings / couplings / wavetubes & d.c. synchronous motors.
Rare Earth Garnets e.g. Ln3Fe5O12 and Y3Fe5O12.
FERRIMAGNETISM shows an unusual temperature-dependence.
- As T increases moment falls to zero at the Condensation Temperature.
- Above Condensation Temperature moment rises in the opposite direction to a maximum.
- Moment, then falls to zero at the Curie Temperature in the normal manner.
Reason?
- The magnetic moments of the rare earth and iron ions oppose each other.
- The rare earth moments dominate at low temperature.
- The rare earth moments randomize at a lower temperature than the iron moments.
Electronic Spectroscopy
Transitions which involve only a redistribution of electrons within the 4f orbitals (f-f transitions) are orbitally-forbidden by the Selection Rules.
Pale colours of LnIII compounds are usually not very intense.
Crystal/Ligand field effects in lanthanide 4f orbitals are virtually insignificant.
4f electrons are well shielded from external charge by 5s2 & 5p6 shells.
f-f absorption bands are very sharp (useful fingerprinting and quantisation of LnIII).
[ d↔d transitions in transition metal compounds are also orbitally forbidden, but gain intensity from and are broadened by the effects of molecular vibrations in distorting the crystal field ].
Optical spectra are virtually independent of environment - similar spectra in gas/solution/solid (sharp lines like typical gas atom spectra).
Insensitivity of f-f transitions means of limited use in study of lanthanide materials.
CeIII and TbIII have high intensity bands in the UV due to 4fn →4fn-15d1 transitions, i.e. f-d and therefore not orbitally forbidden.
Fluorescence / Luminescence
Of certain lanthanides e.g. Tb, Ho & Eu.
Luminescence: emission of light by material from its absorbing energy.
Photoluminescence: use of photons for excitation.
Fluorescence: short time lapse (~ 10-8s) between excitation & emission.
Phosphorescence: long decay times - luminescence continues long after excitation source is removed.
Lasers
One of the most common high power lasers is the Neodymium YAG laser.
Host material is Yttrium Aluminium Garnet (YAG), Y3Al5O12, doped with Nd3+.
Solid State Chemistry
Halides
Halides of the form LnX2, LnX3 & LnX4 exist.
LnX4
- Only (Ce, Tb, Pr)F4 are known.
- correlation with I4 of Ln.
- fluorides only - most oxidising halogen.
- CeF4 is comparatively stable e.g. crystallizes as a monohydrate from (aq).
- TbF4, PrF4 are thermally unstable and oxidize H2O, i.e. prepare dry.
- MF4 all white solids with the UF4/ZrF4 structure.
- Dodecahedral coordination of M.
LnX3
All LnX3 are known (except Pm [not attempted] & possibly EuI3).
Typically crystalline / high mp / deliquescent.
Typically obtained as hydrates from (aq).
On heating, react with water to form oxyhalides: LnX3 + H2O → LnOX + 2HX.
At high temperatures react even with glass: 2LnX3 + SiO2 → 2LnOX + SiX4.
Preparation of anhydrous LnX3
LnF3: Ln(NO3)3(aq) + 3HF → LnF3•0.5H2O (very insoluble).
Heat this under an HF atmosphere (for heavy Ln) to obtain anhydrous LnF3.
LnCl3: Ln2O3 / Ln2(CO3)3 + HCl(aq) → LnCl3•6-8H2O (rather soluble).
Heat this under HCl atmosphere (for heavy Ln) to obtain anhydrous LnCl3.
or
Heat at 300°C, Ln2O3 + 6NH4Cl → 2LnCl3 + 3H2O + 6NH3.
LnBr3 / LnI3: Best by direct combination (susceptible to hydrolysis to oxyhalides). Purify by sublimation (but avoid contact with hot silica!)
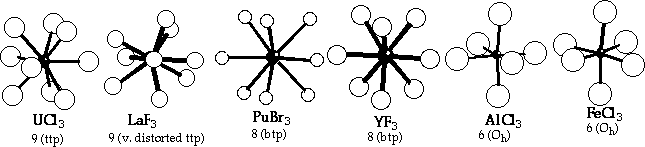
Structures:
Ln coordination varies from 9 for light trifluorides to 6 for heavy iodides.
Coordination Environments from LnX3 structures:
LnX2
Preparation - typically from comproportionation - Ln + 2LnX3 ⇌ 3LnX2
(Sm,Eu,Yb)I2 are obtained from thermal decomposition of LnX3
LnX2 are easily oxidised.
Liberate H2 from H2O [ Except for EuX2 ].
Occurrence of dihalides - parallels high values for I3.
Depends upon the oxidizing power of the halogen (iodides most numerous!)
Trends in the Stability of MX2
Consider:
3MX2(s) ⇌ M(s) + 2MX3(s)
ΔmH° = 3ΔLH(MX2) – 2ΔLH(MX3) + 2I3 - ΔatmH°(M) - (I1 + I2)
Irregularities should follow [ 2I3 - ΔatmH°(M) ], i.e. effectively follow I3 [since variation in -ΔatmH follows closely variation in I3.
This explains occurrence of MCl2:
- La, Ce, Pr MCl2 unknown
- (Sm, Eu, Yb)Cl2 are the most stable MCl2 - may be prepared from LnCl3(s) + ½H2
Structures
- Coordination numbers from 9 to 6
- Fluorides are Fluorite (CaF2) [C.N. = 8]
- Nd,Sm,Eu chlorides are PbCl2-type [C.N. = 7 + 2]
- Nd,Sm,Eu bromides and iodides are SrBr2 type [mixed CN=7 & 8]
- (Dy,Tm,Yb) I2 have layer structures (CdCl2,CdI2) [CN=6] with polarization effects
Two Classes of dihalide
1. Most LnX2 are regarded as "salt-like" halides (insulators).
2. (Ce,Pr,Gd) I2 have metallic lustre, high conductivity.
Formulation as Ln3+(I-)2(e-) with the electron in a delocalized conduction band.
LnII Compounds are finding increasingly more uses .
Lower Halides
LnX3/Ln melts yield phases of reduced halide formulae e.g. Ln2X3 & LnX.
"Reduced Halides" contain "condensed metal clusters".
Black & metallic increase delocalization of electrons through the metal-metal bonded networks.
Gd2X3 single chains of edge-sharing metal octahedra with M6X8-type environment (i.e. face-capped by X).
Lowest Halides are stabilized by H, C or N atoms encapsulated in Ln6 cluster octahedra.
e.g. Gd2Cl2C2 Layers of edge-sharing M6C units
e.g. Gd3Cl3C Framework of M6C units
Hydrides
Preparation: Heat at 300-350°C, Ln + H2 → LnH2
Properties of LnH2
Black, reactive, highly conducting, with fluorite structure.
Most thermodynamically stable of all binary metal hydrides.
Formulated as Ln3+(H-)2(e-) with e- delocalized in a metallic conduction band.
Further H can often be accommodated in interstitial sites, and they are frequently non-stoichiometric, e.g. LuHx where x = 1.83-2.23 & 2.78-3.00.
With a high pressure of H2 forms LnH3.
Reduced conductivity: salt-like Ln3+(H-)3, except for Eu and Yb (the most stable LnII).
Possible Applications of Rare Earth Intermetallic Hydrides.
- Production of ultra-pure hydrogen.
- Isotope Separation of deuterium and hydrogen.
- Source of fuel for motor vehicles.
- Electrodes in Protonic Batteries/Fuel Cells.
- Load Levelling in Power Stations.
- Chemical heat-pump systems.
- Useful hydrogenation agents in organic chemistry.
Binary Oxides
Ln2O3
The most common lanthanide oxide (notable exceptions; CeO2, Pr6O11, Tb4O7).
Sesquioxides, Ln2O3, are strongly basic, and absorb water / CO from air, forming the hydroxide / carbonate salts.
They have pale colours, and properties strongly resemble alkaline earth oxides.
Adopt three structure types:
A-type
- light Ln
- unusual LnO7 capped-octahedra
B-type
- middle Ln
- LnO7 units of three types:
- 2 x capped trigonal prisms
- 1 x capped octahedron
- B-M2O3 structure is very complex. Densest of M2O3 structure types - favoured by increased pressure.
C-type
- heavy Ln
- LnO6 units, but not octahedra
- (face & body - divacant cubic)
- C-type M2O3 is related to Fluorite (MO2) with ¼ of anions removed.
Polymorphism
A (high T)
B (medium T)
C (low T)
Ln(OH)3
Obtained by action of conc. NaOH on Ln2O3 under hydrothermal conditions.
9-coordinate Ln with tricapped trigonal prismatic geometry.
Basicity increases with Z - correlates with decrease in r(Ln3+)
LnO2
CeO2 (most stable).
Fluorite (CaF2) structure.
Pr6O11, Tb4O7 (formed at high p(O2) and high temperature)
Range of non-stoichiometric phases between Fluorite LnO2 and C-type LnO1.5 - intermediate phases were the first known examples of shear structures.
LnO
known for some Ln.
Preparation: Comproportionation Ln + Ln2O3 → 3LnO.
NaCl structure.
NdO, SmO lustrous golden yellow, conducting formulated as Ln3+(O2-)(e-).
EuO (dark red), YbO (greyish-white) insulating genuine Ln2+O2-.
EuO is ferromagnetic and an insulator when pure.
Borides
A variety of binary borides exist e.g. YB2, YB4, YB6, YB12, YB66.
Most important are LnB6 – contain B6 octahedral clusters, and are isomorphous with CaB6.
Black, metallic conductivity (c.f. CaB6 white, insulator).
formulated as Ln3+(B62-)(e-), except EuB6, YbB6 which are Ln2+(B62-).
MT4B4 compounds
M = Sc, Y, Ln, Th, U.
T = Ru, Os, Co, Rh, Ir.
Of current interest for their superconductivity, e.g. CeCo4B4.
Carbides
Class III
Interstitial Carbides.
Close-Packed M with C in Octahedral Interstices, e.g. (Tb,Ho,Y)2C.
anti-CdCl2 structure.
Class II
Occurrence with La – Ho.
M3C C randomly distributed in ⅓ of Octahedral holes in CCP M.
M2C3 Pu2C3 structure - C2 groups.
MC2 CaC2 structure - C2 groups.
Metallic lustre & conductivity - not salt-like.
Hydrolysis of (La,Ce)C2 at ambient temperature gives ethyne, C2H2.
Organometallic Chemistry
Organolanthanide chemistry is not as extensive as organotransition metal chemistry.
Currently receiving a lot of attention, especially in C-H Bond Activation studies.
Primarily ionic in their bonding - contracted nature of the 4f valence orbitals.
Lanthanides cannot act as π-bases hence Ln-CO compounds are not stable.
Organolanthanides are extremely air & moisture sensitive - reflects highly carbanionic character of organic ligand & oxophilicity of Ln2+/3+.
Cyclopentadienides
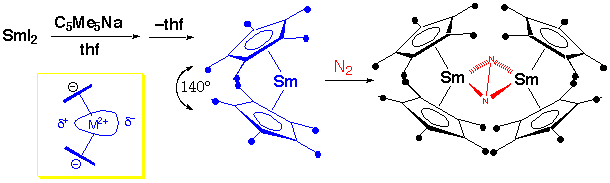
Preparation:
Structure:
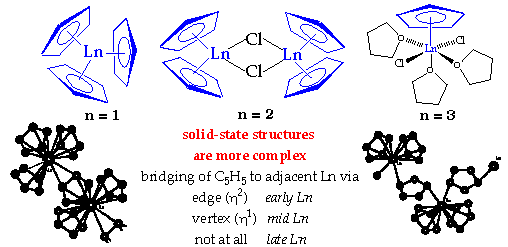
Also in +2 Oxidation State
Alkyls & Aryls
σ-bonded alkyls & aryls.
From metathesis in ether/THF.

R = phenyl probably polymeric.
R = CH2CMe3 stable as LnR3(THF)2.
[LnMe6]3- have been isolated for most Ln.
Mixed Alkyl Cyclopentadienides
C5Me5 (Pentamethyl-cyclopentadienyl) is a common organo-Ln ligand.
Large bulk - only 2 C5Me5 may be bound to Ln.
Causes major change in structural & chemical properties, especially novel chemistry in mixed alkyl cyclopentadienides, e.g. (C5Me5)2LuCH3.
- catalyzes alkene polymerization (Ziegler-Natta chemistry).
- reacts with C-H bonds of extremely low acidity e.g. CH4.
Arenes
Lanthanide Bis-Arene 'sandwich' compounds.
prepared by metal vapour synthesis (MVS) techniques.
stable at ambient temperature.

Ln(0) - such compounds could not be expected for LnIII with contracted 4f orbitals.
Comparisons and Contrasts
Yttrium - Why consider it with the Lanthanides?
- Occurs with lanthanides in rare earth minerals, e.g. monazite.
- Y occurs effectively exclusively in +3 oxidation state.
- combines with non-metals → YHal3 Y2O3, Y3+(H-)2(e-), YH3, etc...
- Y3+ has same radius as Ho3+ and is difficult to separate from it.
- Forms complexes of high coordination number with chelating O-donors, e.g. Y(acac)3(H2O).
- Typical organometallics include: Y(C5H5)3 (polymeric in the solid state).
- Dimeric Y(C5H5)2Cl, monomeric form is a THF adduct
6 Reasons why Scandium could be considered with the Lanthanides
- Sc occurs effectively exclusively in +3 oxidation state.
Combines with non-metals → ScHal3 Sc2O3, etc...
But coordination octahedral (small size).
- Sc forms reduced halides.
e.g. Sc7Cl12 which is Sc3+(Sc6Cl12)3- with Sc6 clusters (but c.f. Nb).
- Scandium Hydride ScH2 is highly conducting Sc3+(H-)2(e-).
- Forms complexes of high coordination number with chelating O-donors, but forms octahedral complexes with monodentate ligands.
- Nitrate & Sulphate are obtained as hydrated salts.
- Typical organometallics include: Sc(C5H5)3 (polymeric in the solid state). Dimeric Sc(C5H5)2Cl, monomeric form is a THF adduct.
4 Reasons why Scandium could be considered as main group 3
- Sc3+ (r = 74 pm) is appreciably smaller than any of the rare earths - behaviour intermediate between the Lanthanides & Aluminium.
- Sc2O3 is more like Al2O3 than Ln2O3: amphoteric in excess OH-.
- ScF3 dissolves in excess F- (N.B. scarcity of halogeno complexes for Lanthanides).
- Anhydrous ScCl3 is easily obtained by P2O5-dehydration of hydrated halide, but unlike AlCl3, ScCl3 is not a Friedel-Crafts catalyst.
Scandium may with similarly few exceptions be viewed as a 1st Row Transition Metal
Six-coordinate complexes are typical.
Aqua-ion is Sc(H2O)63+ and is susceptible to hydrolysis → O-H-bridged dimers...
Some CONTRASTS between Lanthanides & Pre-Transition & Transition Metals
|
Pre-Transition Metals |
Lanthanides |
Transition Metals |
|
Essentially Monovalent - show Group (n+) oxidation state |
Essentially Monovalent (+3). +2/+4 for certain configs |
Show Variable Valence (extensive redox chemistry) control by environment - ligands, pH etc… |
|
Periodic trends dominated by (effective nuclear) charge at noble gas config (i.e. on group valence). |
Lanthanide Contraction of Ln3+. |
Size changes of Mn+ less marked. |
|
Similar Properties for a given group (differentiated by size). |
Similar Properties (differentiated by size). |
Substantial Gradation in Properties. |
|
widespread on earth. |
common mineralogy |
diverse mineralogy |
|
No Ligand Field Effects. |
Insignificant Ligand Field Effects. |
Substantial Ligand Field Effects. |
|
Always 'hard' (O, Hal, N donors) (preferably -vely charged) |
Always 'hard' (O, X, N donors) (preferably -vely charged) |
Later (increasingly from Fe-Cu)/heavier metals may show a 'soft' side. |
|
'Ionic' or 'Covalent' Organometallics |
'Ionic' Organometallics. |
'Covalent' Organometallics. |
|
No Ligand Effects. |
Paucity of Ligand Effects. |
π-Acceptor Ligands - Extensive Chemistry. |
|
Poor Coordination Properties (C.N. determined by size). |
High Coordination Numbers (C.N. determined by size). |
Extensive Coordination C.N. = 6 is typical maximum (but many exceptions). |
|
Flexibility in Geometry. |
Flexibility in Geometry. |
Fixed (by Ligand Field effects) Geometries. |
|
No Magnetism from the metal ions - noble gas configurations of ions |
Free Ion-like Magnetism ground state magnetism |
Orbital Magnetism 'Quenched' by Ligand Fields. excited J-states populated. |
|
'Ionic' compound formulations → large HOMO-LUMO gaps → UV CT spectra |
Weak, Narrow Optical Spectra. Forbidden, unfacilitated transitions. |
Stronger, Broader Optical Spectra. Forbidden transitions. Vibronically-assisted. |
The Actinides
Naturally Occurring Actinides
- Only Actinium, Thorium, Protactinium & Uranium occur naturally (i.e. Z ≤ 92).
- Actinium & Protactinium occur only in trace amounts.
- Neptunium & Plutonium occur in uranium minerals in minute amounts - not appreciated until after they had been synthesised that the synthesis route might occur naturally!
- All isotopes of all the actinides are radioactive.
- Most of the longer-lived isotopes decay by α-emission.
- Both Thorium and Uranium are far from rare.
Thorium
- Widely dispersed, accounts for > 3ppm of the earth's crust.
- Natural Thorium is essentially 100% 232Th.
- Occurs in monazite [with the rare earths] and in uranothorite [a mixed Th, U silicate].
- Obtained as ThO2, thoria, from mineral extraction process.
- Used as 99% ThO2 / 1% CeO2 in thoria gas mantles.
Uranium
- Widely distributed - found scattered in the faults of old igneous rocks.
- Natural Uranium is 99.27% 238U & 0.72% 235U.
- Obtained usually as UO2.
- Used for nuclear fuel, and on a smaller scale for colouring glass/ceramics.
Basic Features of Nuclear Structure & Chemistry and Radiochemistry
All Radon isotopes are short half-life α-emitters (but give rise to short-lived β-emitters). Radon gas is derived from Thorium content in granite minerals - hazard in igneous areas.
Actinium and Protactinium occur in uranium ores in trace amounts, because of their participation in Actinium Decay series (from 235U).
Synthesis of Trans-Uranium Elements – bombardment techniques.
Are there any uses for trans-Uranium elements?
Plutonium:
- 239Pu - produced from 238U by neutron capture in all nuclear reactors.
- Acts as nuclear fuel in fast-breeder reactors.
- Processed for nuclear weapon applications.
- Used as a compact energy source due to the heat from α-decay.
- N.B. α-emission is harmless, unless the emitter is ingested.
- Deep-sea diving suits are heated by ca. 750g of 238Pu
- Combined with PbTe thermoelectric - totally reliable electricity.
- Used in Apollo space missions.
- Human heart pacemakers.
Americium:
- 241Am is used as the α-emission source in smoke alarms
Actinide Metals
Preparation
General method for all Actinides:
Reduction of AnF3 or AnF4 with vapours of Li, Mg, Ca or Ba at 1100 - 1400°C
Highly Electropositive.
Typically react with:
- air → tarnishing
- boiling water or dilute acid → releasing Hydrogen
- most non-metals in direct combination
Structures
Very dense metals (e.g. U = 19 g cm-3) with distinctive structures, e.g. Plutonium has at least 6 allotropes and forms numerous alloys.
General Observations (comparisons with Lanthanides)
- Electronic Configurations of Actinides are not always easy to confirm.
- Atomic spectra of heavy elements are very difficult to interpret in terms of configuration. Competition between 5fn7s2 and 5fn-16d7s2 configurations is of interest.
- For early actinides promotion 5f → 6d occurs to provide more bonding electrons. Much easier than corresponding 4f → 5d promotion in lanthanides.
- Second half of actinide series resemble lanthanides more closely.
- 5f orbitals have greater extension wrt 7s and 7p than do 4f relative to 6s and 6p orbitals, e.g. ESR evidence for covalent bonding contribution in UF3, but not in NdF3.
- 5f / 6d / 7s / 7p orbitals are of comparable energies over a range of atomic numbers, especially U–Am.
- Tendency towards variable valency.
- Greater tendency towards (covalent) complex formation than for lanthanides, including complexation with π-bonding ligands.
- Electronic structure of an element in a given oxidation state may vary between compounds and in solution.
- Often impossible to say which orbitals are being utilized in bonding.
- Ionic Radii of ions show a clear "Actinide Contraction"
- Actinide 3+ or 4+ ions with similar radii to their Lanthanide counterparts show similarities in properties that depend upon ionic radius
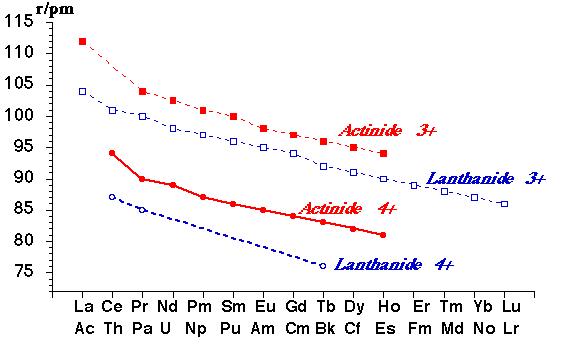
Electronic Spectra
- Narrow bands (compared to transition metal spectra).
- Relatively uninfluenced by ligand field effects.
- Intensities are ca. 10 x those of lanthanide bands.
- Complex to interpret.
Magnetic Properties
- Hard to interpret.
- Spin-orbit coupling is large & Russell-Saunders (L.S) Coupling scheme doesn't work.
- Ligand field effects are expected where 5f orbitals are involved in bonding.
Survey of Actinide Oxidation States
+2
- Unusual oxidation state.
- Common only for the heaviest elements.
- No2+ & Md2+ are more stable than Eu2+.
- Actinide An2+ ions have similar properties to Lanthanide Ln2+ and to Ba2+ ions.
+3
- The most common oxidation state.
- The most stable oxidation state for all trans-Americium elements (except No).
- Of marginal stability for early actinides Th, Pa, U (But: Group oxidation state for Ac).
- General properties resemble Ln3+ and are size-dependent.
- Stability constants of complex formation are similar for same size An3+ & Ln3+.
- Isomorphism is common.
- Later An3+ & Ln3+ must be separated by ion-exchange/solvent extraction.
- Binary Halides, MX3 easily prepared, & easily hydrolysed to MOX.
- Binary Oxides, M2O3 known for Ac, Th and trans-Am elements.
+4
- Principal oxidation state for Th.
- Th4+ chemistry shows resemblance to Zr4+ / Hf4+ - like a transition metal.
- Very important, stable state for Pa, U, Pu.
- Am, Cm, Bk & Cf are increasingly easily reduced - only stable in certain complexes, e.g. Bk4+ is more oxidizing than Ce4+.
- MO2 known from Th to Cf (fluorite structure).
- MF4 are isostructural with lanthanide tetrafluorides.
- MCl4 only known for Th, Pa, U & Np.
- Hydrolysis / Complexation / Disproportionation are all important in (aq).
+5
- Principal state for Pa.
- Pa5+ chemistry resembles that of Nb5+ / Ta5+ - like a transition metal.
- For U, Np, Pu and Am the AnO2+ ion is known (i.e. quite unlike Nb/Ta).
- Comparatively few other An(V) species are known.
- e.g. fluorides, PaF5, NbF5, UF5; fluoro-anions, AnF6-, AnF72-, AnF83-.
- e.g. oxochlorides, PaOCl3, UOCl3; uranates, NaUO3.
+6
- AnO22+ ions are important for U, Np, Pu, Am.
- UO22+ is the most stable.
- Few other compounds e.g. AnF6 (An = U, Np, Pu), UCl6, UOF4 etc..., U(OR)6.
+7
- Only the marginally stable oxo-anions of Np and Pu, e.g. AnO53-.
Actinide Aqueous Chemistry
- Latimer & Frost Diagrams for elements in acid & alkaline (aq) indicate actinides are quite electropositive.
- Pa - Pu show significant redox chemistry, e.g. all 4 oxidation states of Pu can co-exist in appropriate conditions in (aq).
- Stability of high oxidation states peaks at U (Np).
- An3+ is the maximum oxidation state for (Cf)Es – Lr.
- No2+(aq) is especially stable ~ most stable state for No in (aq).
- Redox potentials show strong dependence on pH (data for Ac - Cm).
- High oxidation states are more stable in basic conditions.
- Even at low pH hydrolysis occurs - formation of polymeric ions. When hydrolysis leads to precipitation measurement of potentials is difficult, e.g. Pa5+ hydrolyses easily; potentials that indicate it to be the most stable oxidation state are recorded in presence of F- or C2O42-.
- Tendency to disproportionation is particularly dependent on pH, e.g. at high pH 3Pu4+ + 2H2O ⇌ PuO22+ + 2Pu3+ + 4H+.
- Early actinides have a tendency to form complexes - complex formation influences reduction potentials, e.g. Am4+(aq) only exists when complexed by fluoride (15 M NH4F(aq)).
- Radiation-induced solvent decomposition produces H• and OH• radicals, which lead to reduction of higher oxidation states e.g. Pu V/VI, Am IV/VI
Actinide Stereochemistries
Actinide Stereochemistries show similarities with the Lanthanides. High coordination numbers, e.g. [Th(NO3)6]2- has distorted icosahedral (C.N. = 12) geometry, and C.N. = 8, 9 are very common UF82-, Th(S2CNEt2)4
Distortions from idealised stereochemistries
e.g. PuF62- is not octahedral, but widest range of stereochemistries is for An(IV) rather than An(III), possibly because chemistry of early actinides has received most attention!
Complexes
A wide range of complexes with monodentate and chelating ligands.
Complexing ability:- [M5+] > M4+ > MO22+ > M3+ > MO2+
Geometry may be strongly influenced by covalent bonding effects, e.g. MO22+ unit is always linear → UO2(η2-NO3)2(H2O)2 is hexagonal bipyramidal.
Compounds: Actinide Hydrides, Halides, Oxides, Oxyhalides ...
- For a given oxidation state show similarly diverse C.N. to Lanthanides.
- Different accessible oxidation states - even greater diversity of structure.
- Wide variety of oxidation states of ligands & number of oxidation states.
- Extraordinary range of stereochemistry in actinide complexes and compounds.
Uranium Chemistry
Halides
Fluorides
UF6 - the most important fluoride.
Preparation:
UO2 + 4HF → UF4 + 2H2O.
3UF4 + 2ClF3 → 3UF6 + Cl2.
Properties:
mp. 64°C, vapour pressure = 115 mmHg at 25°C.
Made on a large scale to separate uranium isotopes.
Gas diffusion or centrifugation separates 235UF6 from 238UF6.
Uranium richer in 235U is termed enriched, richer in 238U is depleted.
Powerful fluorinating agent.
Other Fluorides
UF6 + Me3SiCl → Me3SiF + ½Cl2 + UF5 (melts to an electrically-conducting liquid).
UF6 + 2Me3SiCl → 2Me3SiF + Cl2 + UF4 → 500-600°C gives UO2 + CFCl2CFCl2.
Mixed-Valence fluorides such as U2F9 also form.
Reduction of UF4 with ½H2 yields UF3.
Chlorides
UCl4 – is the usual starting material for the synthesis of other U(IV) compounds.
Preparation:
Liquid-phase chlorination of UO3 by refluxing hexachloropropene.
Properties:
Soluble in polar organic solvents & in water.
Forms various adducts (2 - 7 molecules) with O and N donors.
UCl3
Usually encountered as UCl3(thf)x (a rather intractable material).
Unsolvated binary gives its name to the UCl3 structure!
Actinide trihalides form a group with strong similarities (excepting redox behaviour) to the Lanthanides.
UCl6
From chlorination of U3O8 + C.
Highly oxidising.
Moisture-sensitive : UCl6 + 2H2O → UO2Cl2 (Uranyl Chloride) + 4HCl.
In CH2Cl2 solution UCl6 decomposes to U2Cl10 (Mo2Cl10 structure).
Halogeno Complexes
All Halides can form halogeno complexes, but F- and Cl- are best-known.
Occurrence:
U(III): UCl52-, U2Cl72- and UCl4-.
U(IV): UF73- and UF84- are common, UF62- and UCl62- are also known. Also pseudohalide complexes, e.g. [U(NCS)8]4-.
U(V): U(V) is usually unstable in (aq), but UF5 in 48% HF → M+UF6- (M+ = Rb+, Cs+, H3O+) salts U(VI): UF7- and UF82- are known, the latter is more thermally-stable.
Hydrides
Principal Uranium Hydride is UH3 – important as a source material for U(III) and U(IV) chemistry.
Oxides
Many binary phases UOx have been reported.
Many are not genuine phases.
Genuine phases show range of O-content.
The most important genuine phases are UO2, U4O9, U3O8, UO3.
UO2 & U4O9
UO2 (black-brown) has the Fluorite structure. Stoichiometric material is best obtained from:

Interstitial Oxide Ions may be incorporated into the structure - UO2+x.
Neutron Diffraction studies indicate oxide vacancies in the normal fluorite lattice.
At UO2.25 (U4O9) (black) - interstitials are ordered forming a distinct phase in the phase diagram.
U3O8 & UO3
U3O8 is dark green.
conveniently made by heating uranyl nitrate or ethanoate in air.
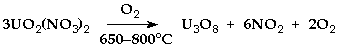
> 650°C Higher uranium oxides decompose to U3O8.
> 800°C loses U3O8 oxygen.
Structure:
Mixed oxide - average oxidation state U5.33.
Evidence suggests Class II/III mixed valence.
All U atoms have essentially identical environments.
Contains pentagonal bipyramidal UO7 units.
UO3 is orange-yellow.
Produced by a variety of methods:
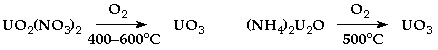
Structure:
> 6 modifications have been characterised.
Most contain O=U=O 'uranyl' groups linked by 4x equatorial bridging O - distorted octahedral environments.
Uranates
Fusion of uranium oxides with alkali or alkaline earth carbonates – orange/yellow/brown mixed-oxides, Uranates.
Aqueous Chemistry
Complex aqueous chemistry due to extensive possibilities for complexation, hydrolytic reactions, often leading to polymeric ion species.
Reduction Potentials appropriate for 1M HClO4 indicate:
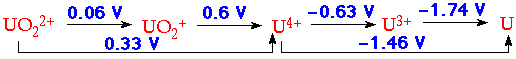
U3+
Powerful reducing agent, reduces H2O to H2 (solutions in 1M HCl stable for days).
Obtained by reduction of UO22+ electrolytically or with Zn/Hg.
U4+
Only slightly hydrolysed in 1M acid solution U4+ + H2O ⇌U(OH)3+ + H+.
But, it can give rise to polymeric species in less acid solutions.
Regarded as a 'stable' oxidation state of uranium in (aq).
UO2+
Extremely unstable to disproportionation.
Evidence for its existence in (aq) from stopped-flow techniques.
More stable in DMSO (half-life ~ 30 mins).
UO22+
The Uranyl ion.
Very stable, and forms many complexes.
A dominant feature of uranium chemistry.
Reduced to U4+ by e.g. Zinc, Cr2+
Re-oxidation by H218O2 → U18O22+
Re-oxidation by 18O2 → U(18O16O)2+
Linear, symmetrical (O=U=O)2+ structure.
Why is it trans linear, whereas WO22+ is cis, bent?
WO22+ (6d0) is cis bent because it allows π-donation from the 2 O to 2 independent d-orbitals, with a single d-orbital shared.
ThO2 (6d05f0) is bent (122o) for similar reasons i.e. no f-orbital participation.
UO22+ (6d05f0) is trans, linear because of the participation of its 5f orbitals.
U(5f) are of considerably lower energy than Th(5f).

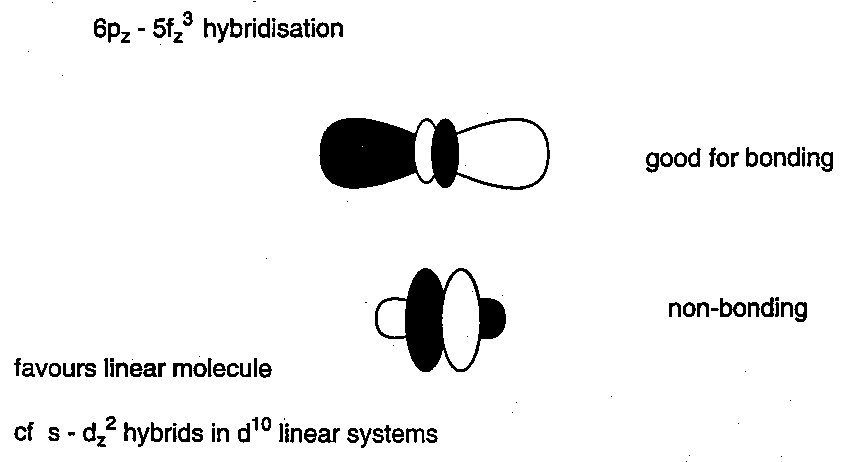
Details of the MO diagram for AnO22+ are controversial, but f-orbitals have ungerade symmetry, d-orbitals are gerade → no d-f mixing in centrosymmetric AnO22+ unit.
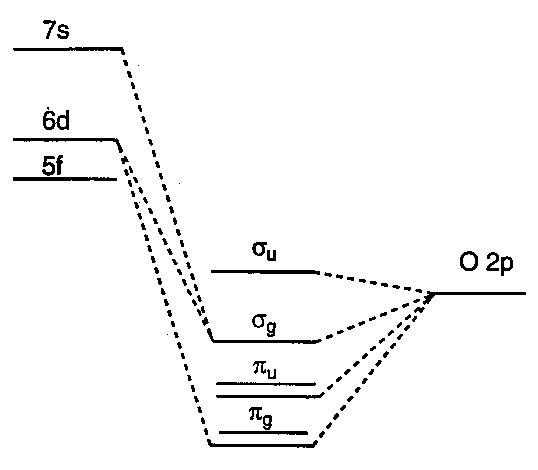
UO22+ readily adds 4-6 donors in its equatorial plane → distinctive complexes
e.g. cyclic hexadentates.
UO22+ salts show characteristic (yellow) fluorescence.
Organometallics
Organometallic chemistry of actinides is relatively recent.
Similar to lanthanides in range of cyclopentadienides / cyclo-octatetraenides / alkyls
Cyclopentadienides are π-bonded to actinides.
Compounds include:-
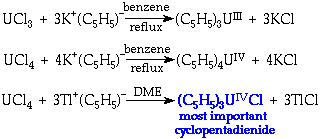
C5H5- does not behave ionically, but Cl- is labile → formation of a wide variety of (C5H5)3UX compounds.
The most notable Cyclooctatetraenide is Uranocene

{kind=link}
- Green crystals, paramagnetic and pyrophoric.
- Stable to hydrolysis.
- Planar 'sandwich'.
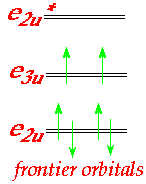
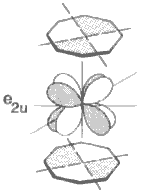
- Eclipsed D8h conformation.
- UV-PES studies show that bonding in uranocene has 5f & 6d contributions.
- e2u symmetry interaction shown can only occur via f-orbitals.
Nuclear Reactors, Atomic Energy & Uranium Chemistry
Principles of Nuclear Reactors
- Nuclear fission = large nucleus splitting into 2 highly energetic smaller nuclei + neutrons.
- Sufficient neutrons of suitable energy can induce fission of further nuclei → Chain reaction.
- To sustain chain reaction a critical mass of uranium must be achieved (prevents neutron loss).
- Kinetic energy of main fragments is converted to heat (106x energy of same mass of coal).
- Only naturally-occurring fissile nucleus is 235U (0.72% natural abundance).
Modern Nuclear Reactors
- Current nuclear reactors use UO2 fuel – less reactive than U metal.
- Enrichment is by fractional gaseous centrifugation of UF6 (easily sublimed).
- Neutron capture by 238U results in formation of 239Pu, which is fissile. Significant amounts of Pu will only be produced in an unmoderated reactor (fuel reprocessing more dangerous!)
Coolants
- Water/Heavy Water – to keep it liquid it must be pressurized.
- CO2 gas – in the Advanced Gas-Cooled Reactor.
I am happy for them to be reproduced, but please include credit to this website if you're putting them somewhere public please!