Free Radicals
Covers generation through reaction types and mechanism onto synthetic uses. I did this topic at Advanced level so should be fairly comprehensive. Mixture of Primer and Lecture Notes.
Free Radicals Notes
Definitions
Free radical – organic species with an unpaired electron, not including carbenes and certain photochemically excited compounds.
Stabilised – include carbon-centred radicals, R•, for which the R-H bond strength is less than that for the corresponding C-H in (1o/2o/3o) alkane. Usually transient.
Persistent – radicals with a lifetime significantly greater than methyl under the same conditions. Not necessarily stabilised. Lifetimes range from seconds to years.
Examples:
|
Non-stabilised, persistent |
|
|
Stabilised, persistent |
|
|
Transient, non-stabilised |
Me• |
|
Transient, stabilised |
Me3C•, allyl• |
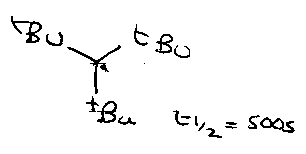

Valence Bond Picture of Heteroatom Stabilisation
Dative:
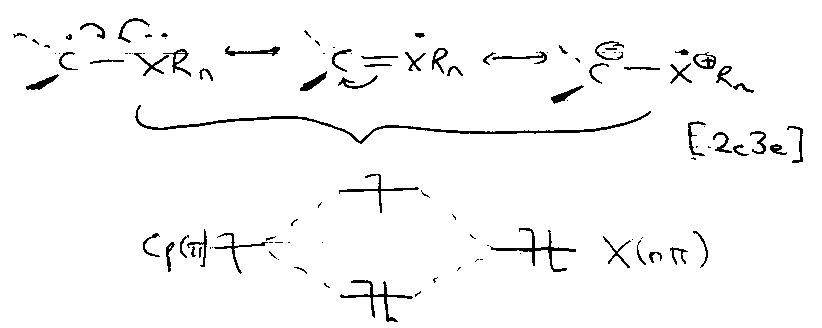
Therefore stabilised.
Capto:
Captodative:
Only radicals can do this (ionic → one way is destabilised).
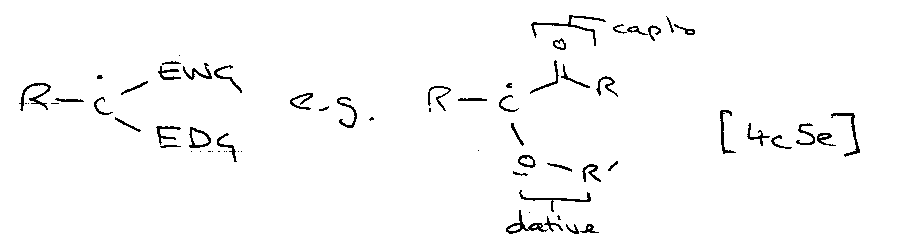
Generation of Free Radicals
Thermal cleavage of weak bonds
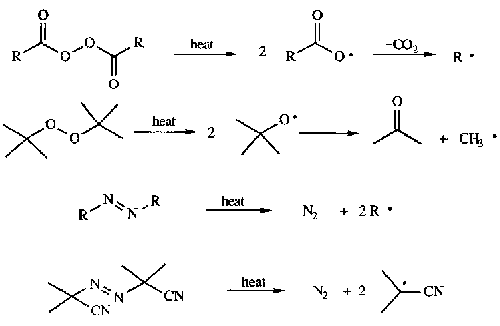
Photochemical cleavage of weak bonds
Useful as heat can destabilise compound.
Initiating compound must absorb light of appropriate wavelength to cleave the bond.
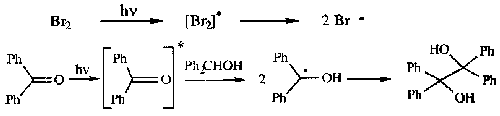
Note that the latter reaction is technically a “phototransformation”.
Electron Transfer
Electrolytic –
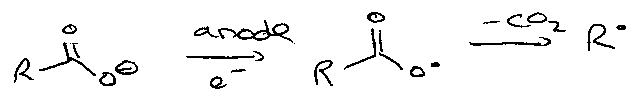
Redox (Fenton) –

Dissolving Metal –
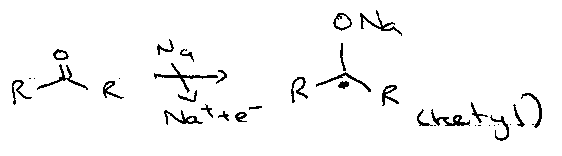
SRN’ (Sandmeyer) –
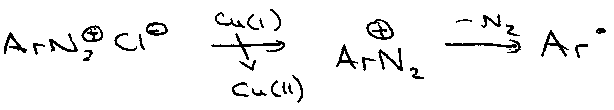
Giese’s Mercury Method –
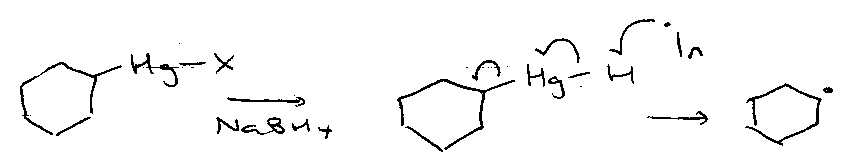
- Room temperature.
- No UV initiation.
- Clean - Hg• by-product.
But, competing direction reduction is bad:
R-Hg-H + R• → RH + Hg•
Birch Reduction –
1 electron process.
Regiochemistry applies when substituted.
See other notes (e.g. Oxidation and Reduction Notes) for the mechanism.
Simple Reactions
Radical Combination –
Combining two radicals to terminate a chain reaction, R•+R• → R-R. Often slow due to low concentrations of radicals generally. Only really viable for long-lived radicals or via solvent cages (can be rapid in the latter case).
Radical Abstraction –
Attack often on a H atom or Halogen atom.
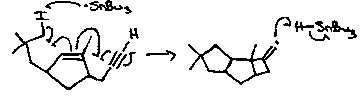
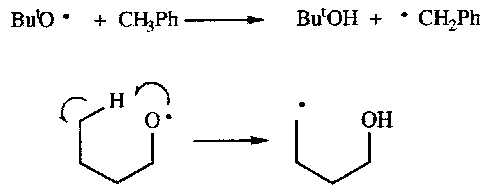
Can be thought of as displacement reaction.
Radical Addition –
Radicals can add to double or triple bonds, and it is often the case that anti-Markovnikov product can be obtained by this route. For example:
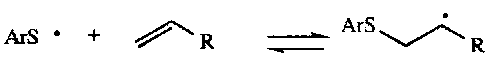
Fragmentation –
This is the reverse of radical addition, and often occurs as β-elimination. An example would be:
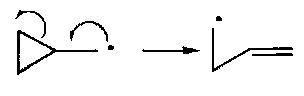
Rearrangements –
These are radical reactions that occur intramolecularly (often abstraction). It can lead to cyclisation of long chain compounds.
Some examples:
Chain Reactions
Example:
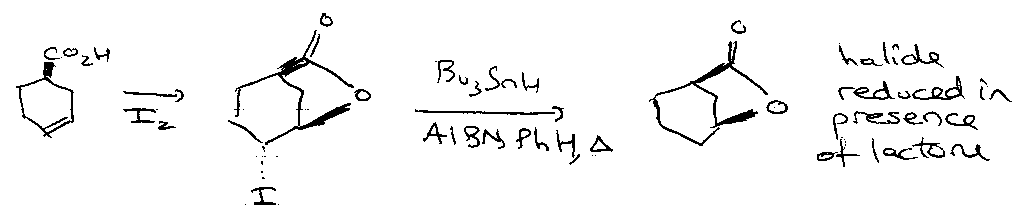
Series of events:
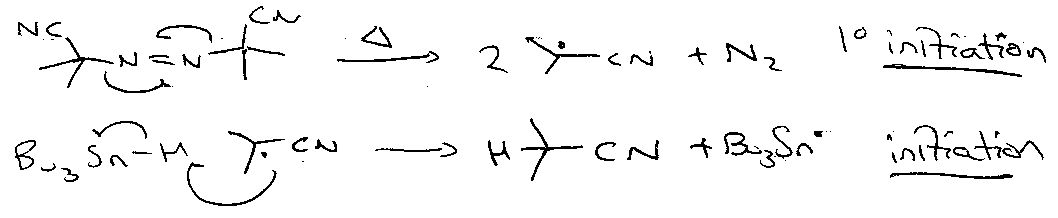
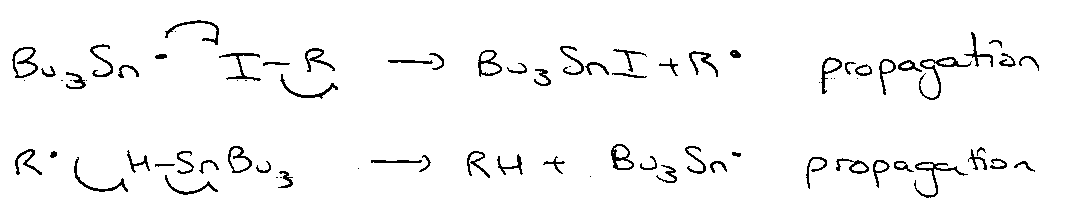
Termination:
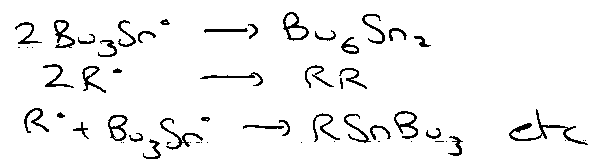
At low concentration, this is unlikely. Minimising termination thus involves:
- Low concentration of radicals → steady initiation (AIBN).
- Fast propagation → 2 x weak bonds to give 2 x strong bonds (e.g. Sn-H + R-I → Sn-I + R-H).
Note that sometimes chain reactions do not occur, particularly with stable radicals or those that are trapped in solvent cages, for example:
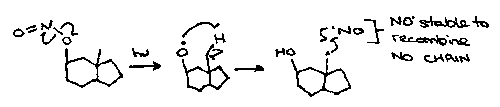
Cyclic Representation –

Radicals Ions & the SET Mechanism
Examples of reactions involving radical ions – pinacol, acyloin (see other notes).
SRN1 Mechanism:
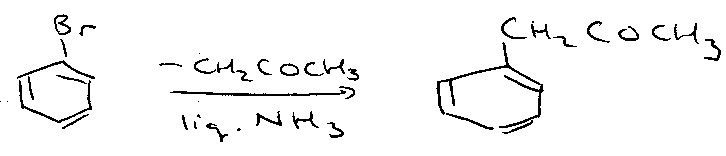
Chain propagation –
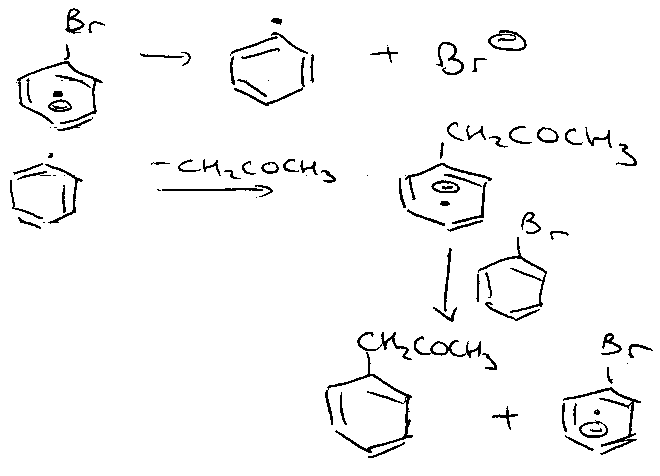
This is another example of Single Electron Transfer, then the SRN1 mechanism:
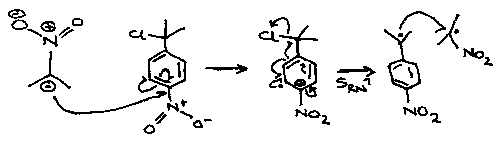
Biradicals and Radical Pairs
e.g.
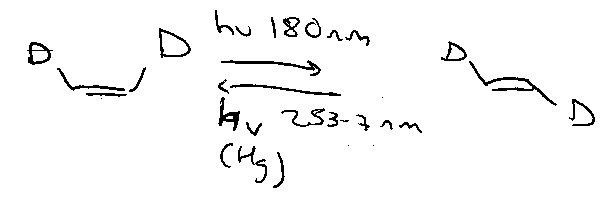
Also, benzene is a biradical in the triplet state.
Example reactions – Wittig & Stevens Rearrangements (see Rearrangements Notes).
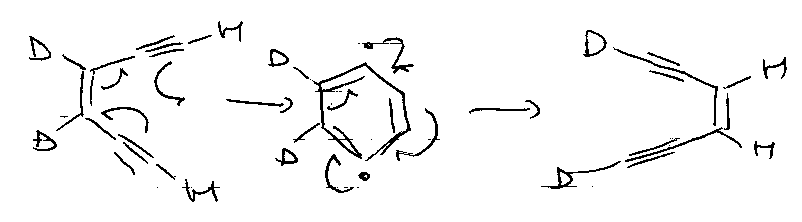
Also, Bergman Cyclisation –
Free Radical Substitution
Homolytic bimolecular substitution. Most common is H-abstraction.
RBr + Bu3SnH → RH + Bu3SnBr
Generally,
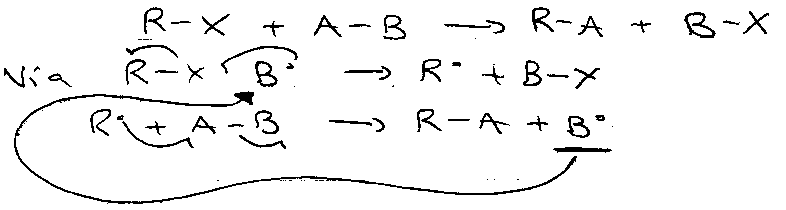
In cyclic form:
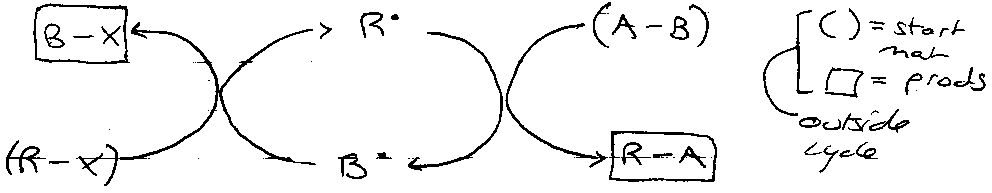
Comparison with (ionic) nucleophilic substitution –
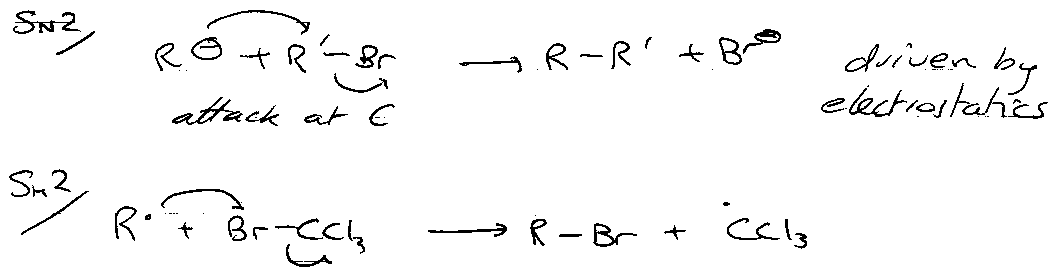
Attack at halogen instead. Driven by availability of the atom and bond strengths (particularly breaking).
Determination of Reactivity and Regioselectivity
Bond Breaking
k = A e-E/RT
ln A ≈ same values for different R in CH3• + H-R → CH4 + R•.
Thus, similar entropy for bimolecular process. Hence, Ea is proportional to D(R-H).
ln k is also proportional to D(R-H) as a result [ linear correlation ], which implies bond breaking is important in the rate determining step.
Bond Forming
ln A similar, so Ea is proportional to D(X-H). Dominates as in bond breaking.
ln k vs. D(X-H) is non-linear, i.e. bond forming may or may not be important → early or late Transition States (Hammond Postulate).
Hammond Postulate –

Thus, for H• abstraction by X•, when X = F reaction is very exothermic → early transition state (like starting materials), therefore little R-H breaking. Hence, T. State is not greatly affected by R:


R-H--------F
When X = Br reaction is endothermic → late transition state (like products). Thus significant R-H breaking. T. State will be sensitive to the nature of R:
R--------H-Br
In general, halogens become less discriminating in H-abstraction in the order:
I > Br > Cl > F
e.g.
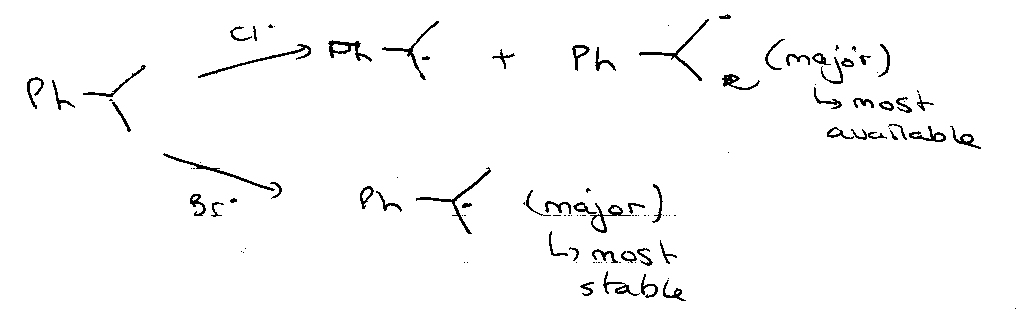
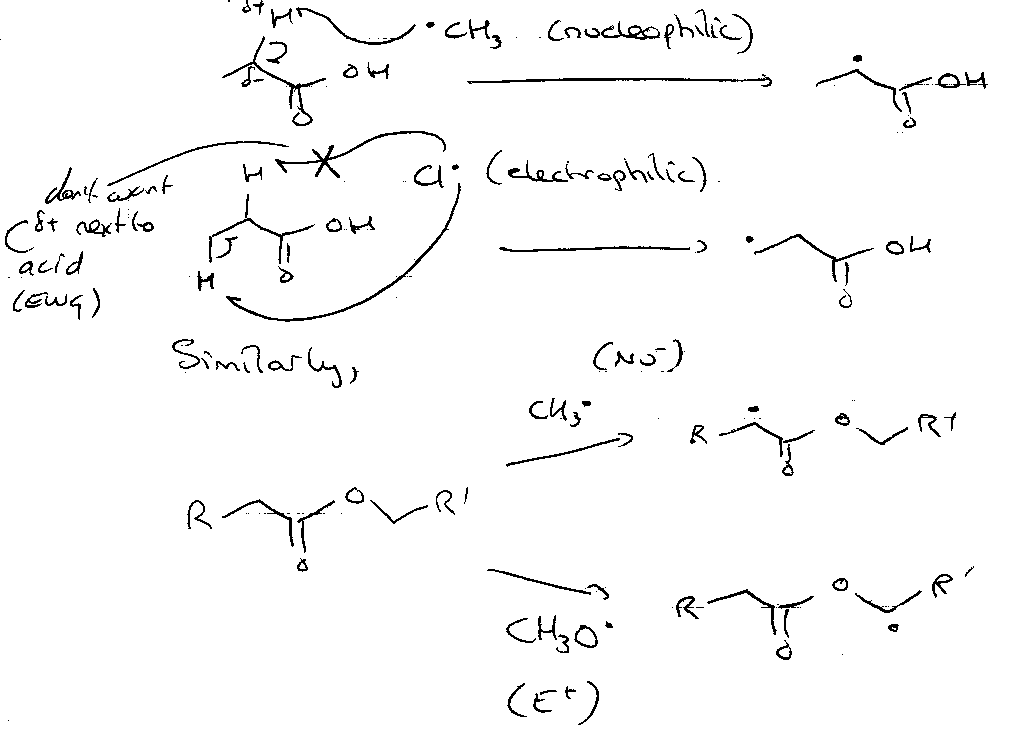
Polarity Effects
Nucleophilic:
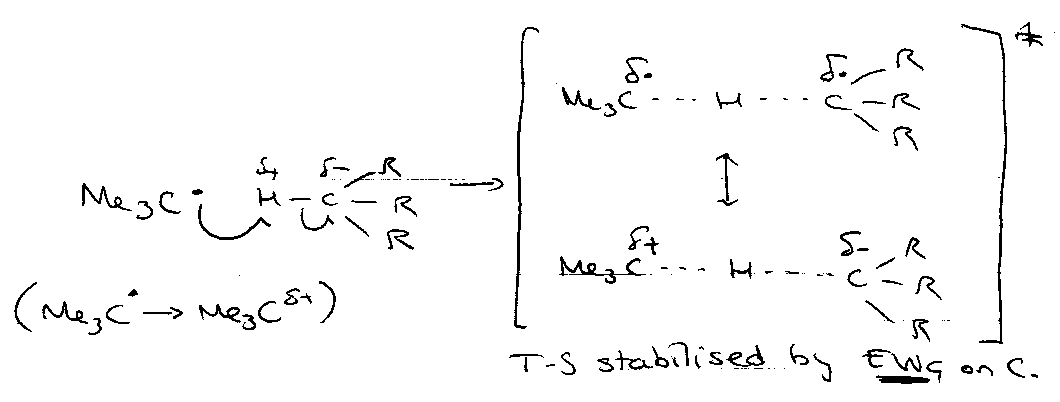
Electrophilic:
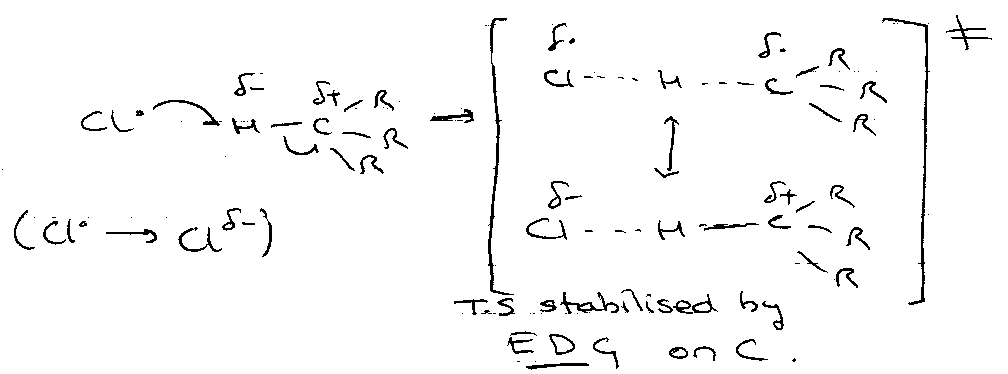
Explains the position of H-abstractions, e.g.
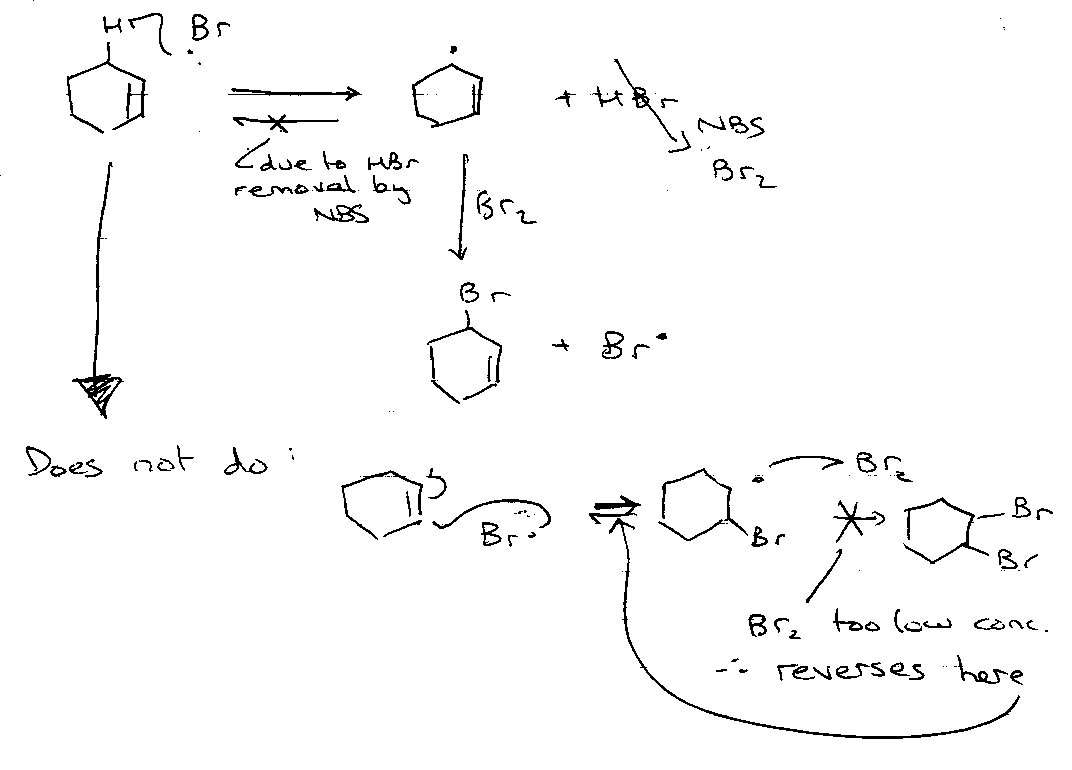
Wohl-Ziegler Allylic Bromination
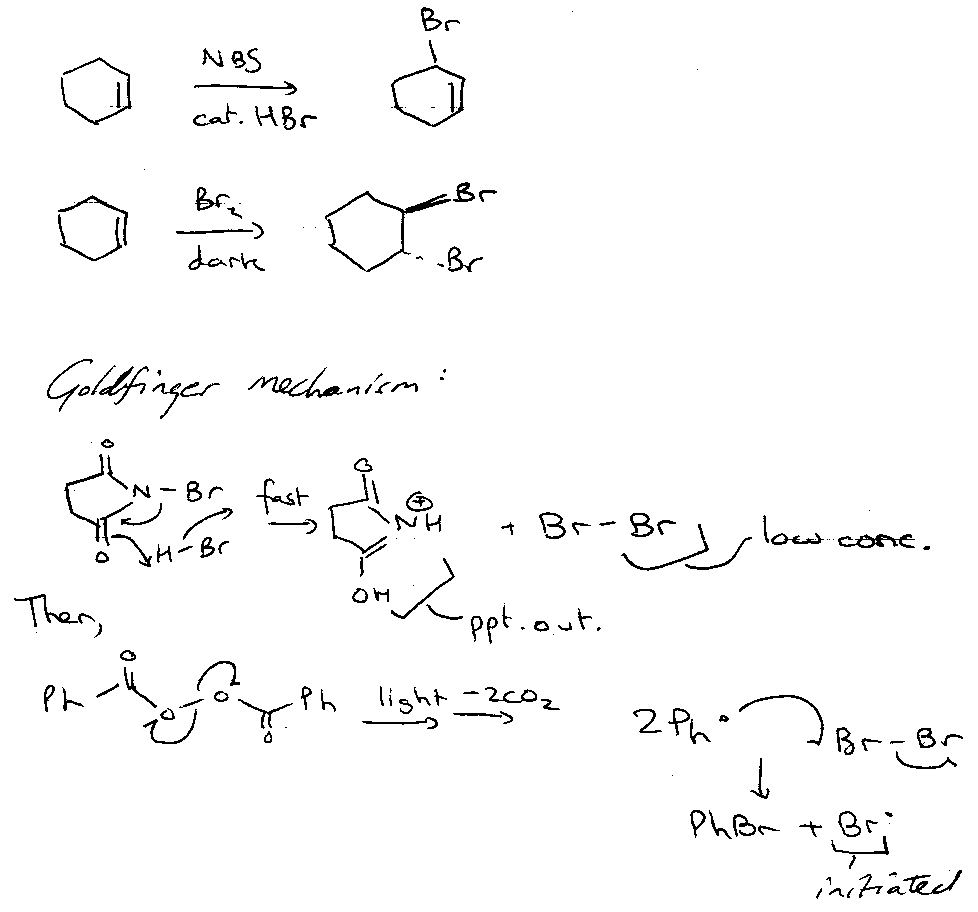
Then,
Addition to Multiple Bonds
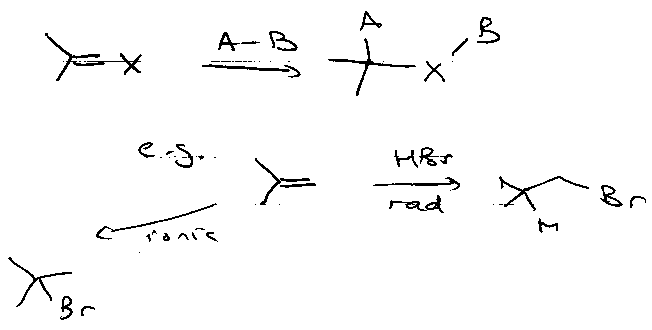
Consider:
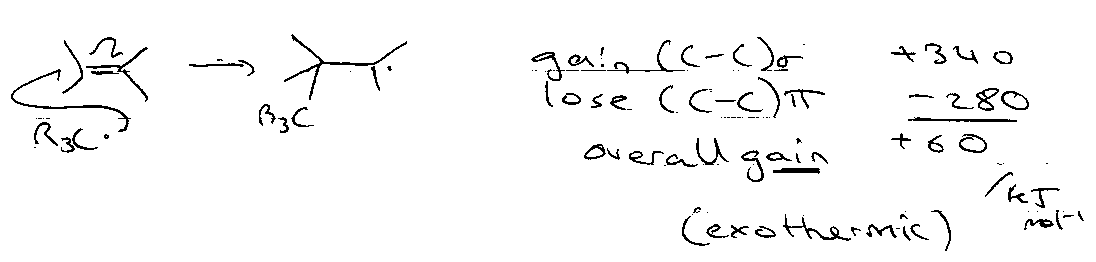
But,
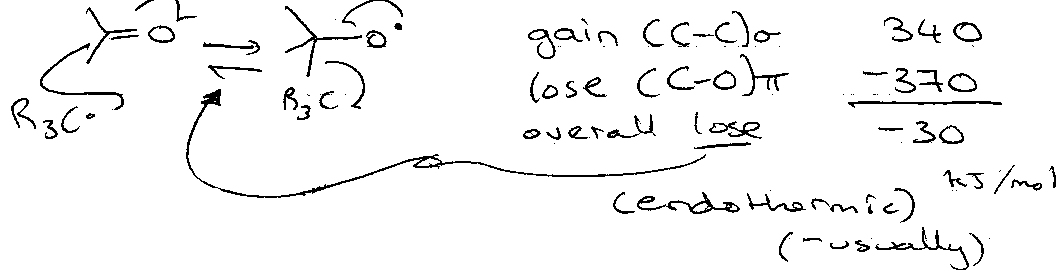
Selectivity
Consider:
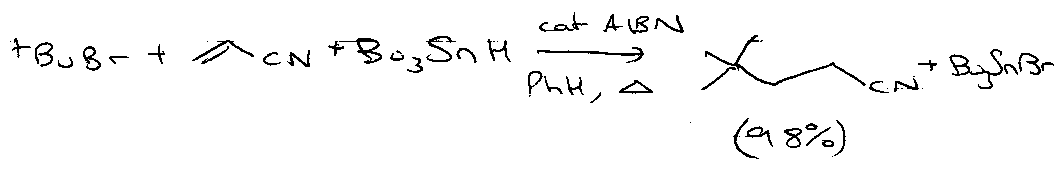
For this to occur:
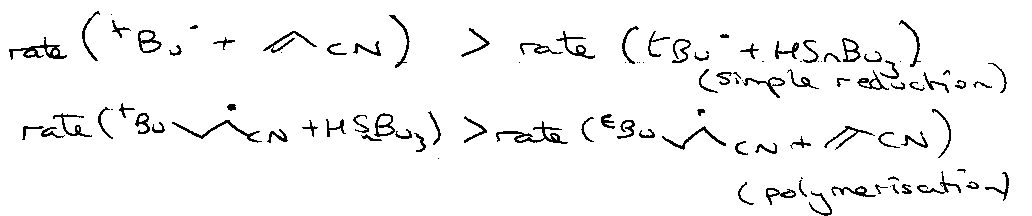
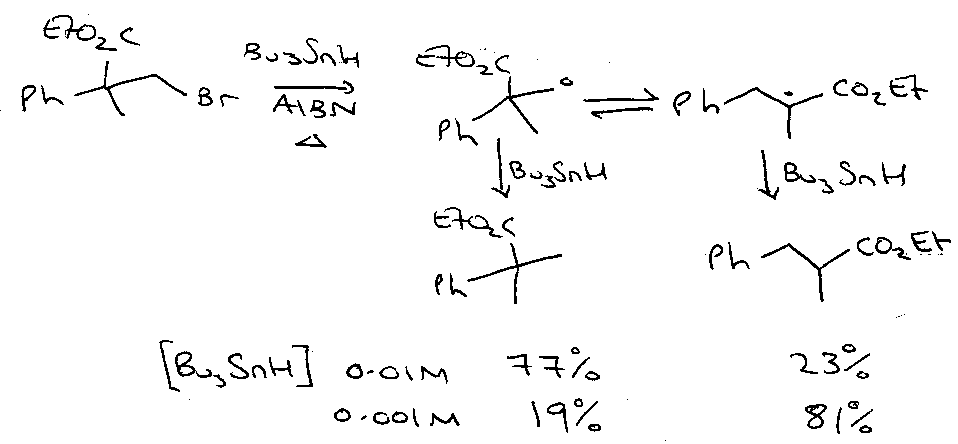
tBu• + HSnBu3 is very fast (3x105 M-1 s-1) so set [Bu3SnH] to be as low as possible. Also set [H2C=CHCN] as high as possible.
Reason why there’s no polymerisation? See below.
Substituent Effects
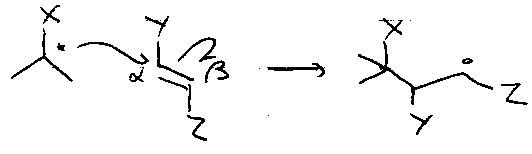
β-effects
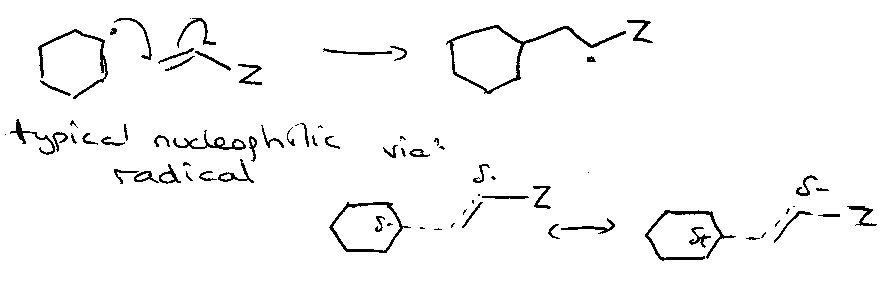
i.e. want electron withdrawing Z to stabilise T. State when nucleophilic radical.
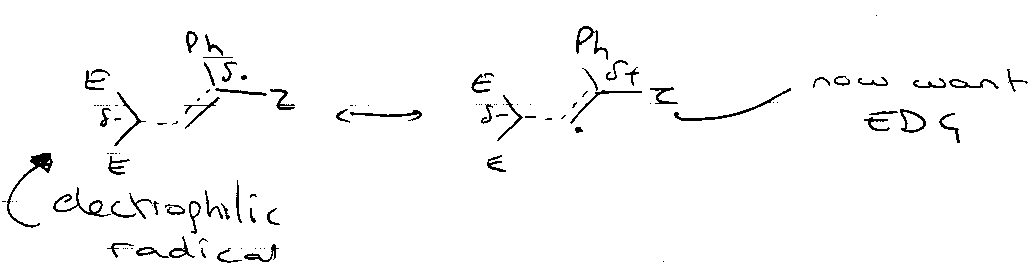
Consider:
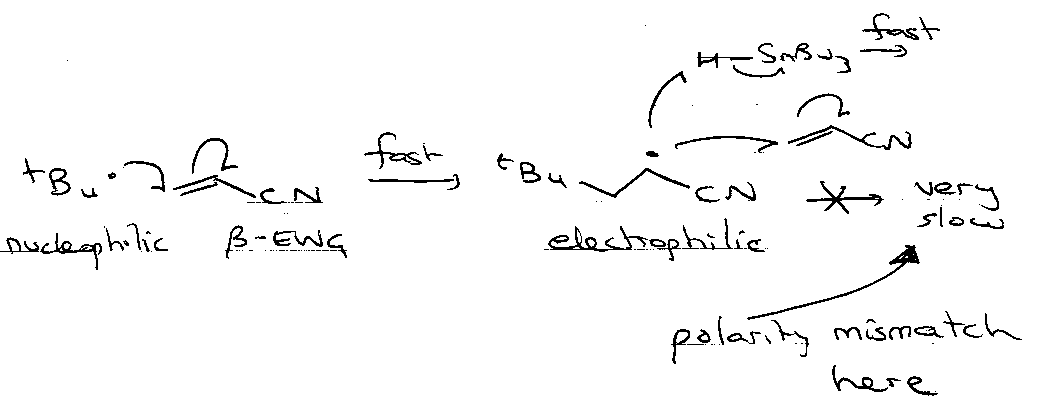
α-effects

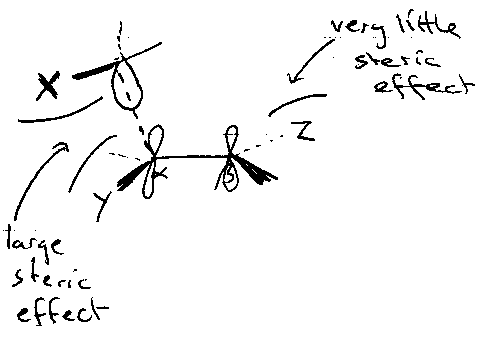
Electron withdrawing group Y favours Nucleophilic radical a little, but sterics usually the dominant factor – often offset any electronically favourable effects operating.
Radical Substituents
Comparable to α-effects – generally slow addition.
Mechanism
Exothermic → early transition state (Hammond Postulate).
Molecular Orbital Description –
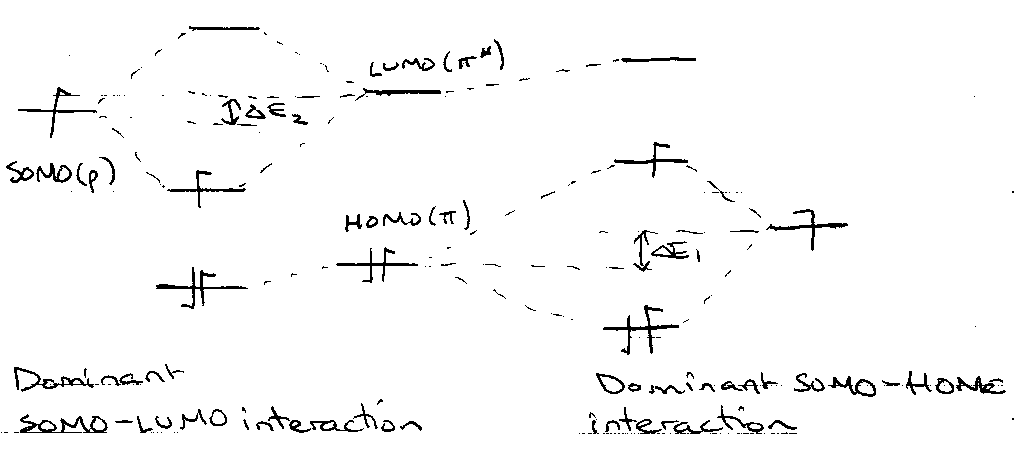
EWG on alkene: lower HOMO and LUMO → ΔE2 < ΔE1.
EDG on alkene: raise HOMO and LUMO → ΔE1 < ΔE2.
Radical Copolymerisation

Allyl Transfer
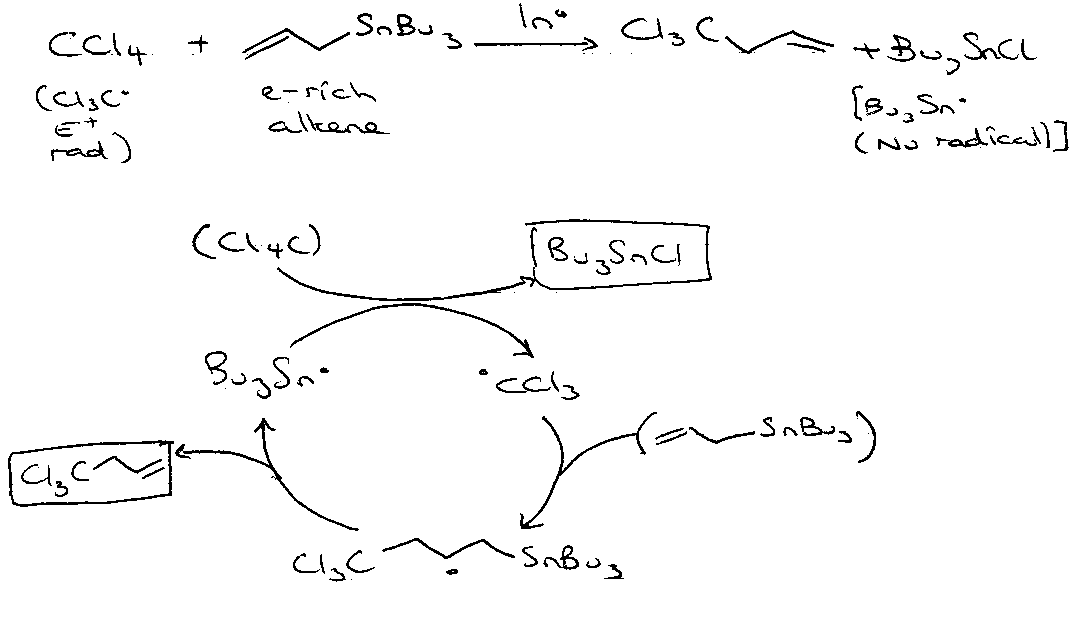
Rearrangements
R• → R’• without change of molecular formula.
e.g. [1,5] H-transfer:
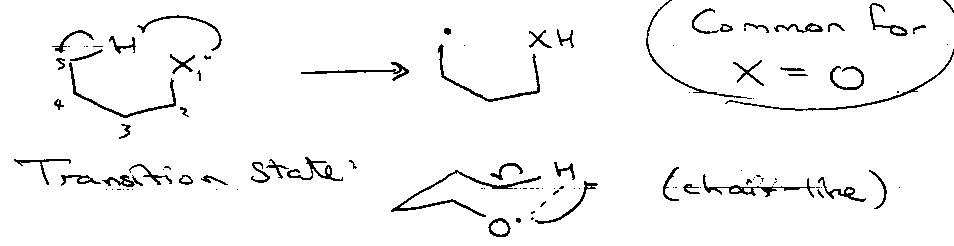
Favourable by ~50-75 kJ mol-1.
Mechanism
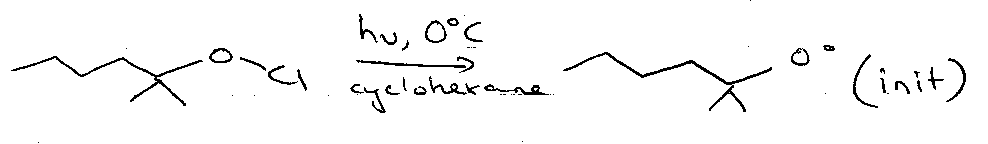
Then several paths available:
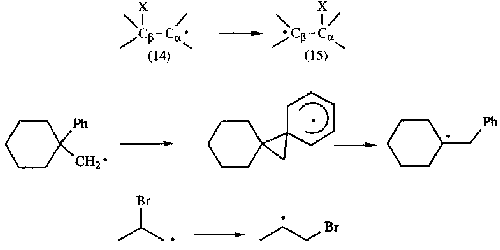
Homobenzylic Rearrangement
(1,2-phenyl shift).
Via:

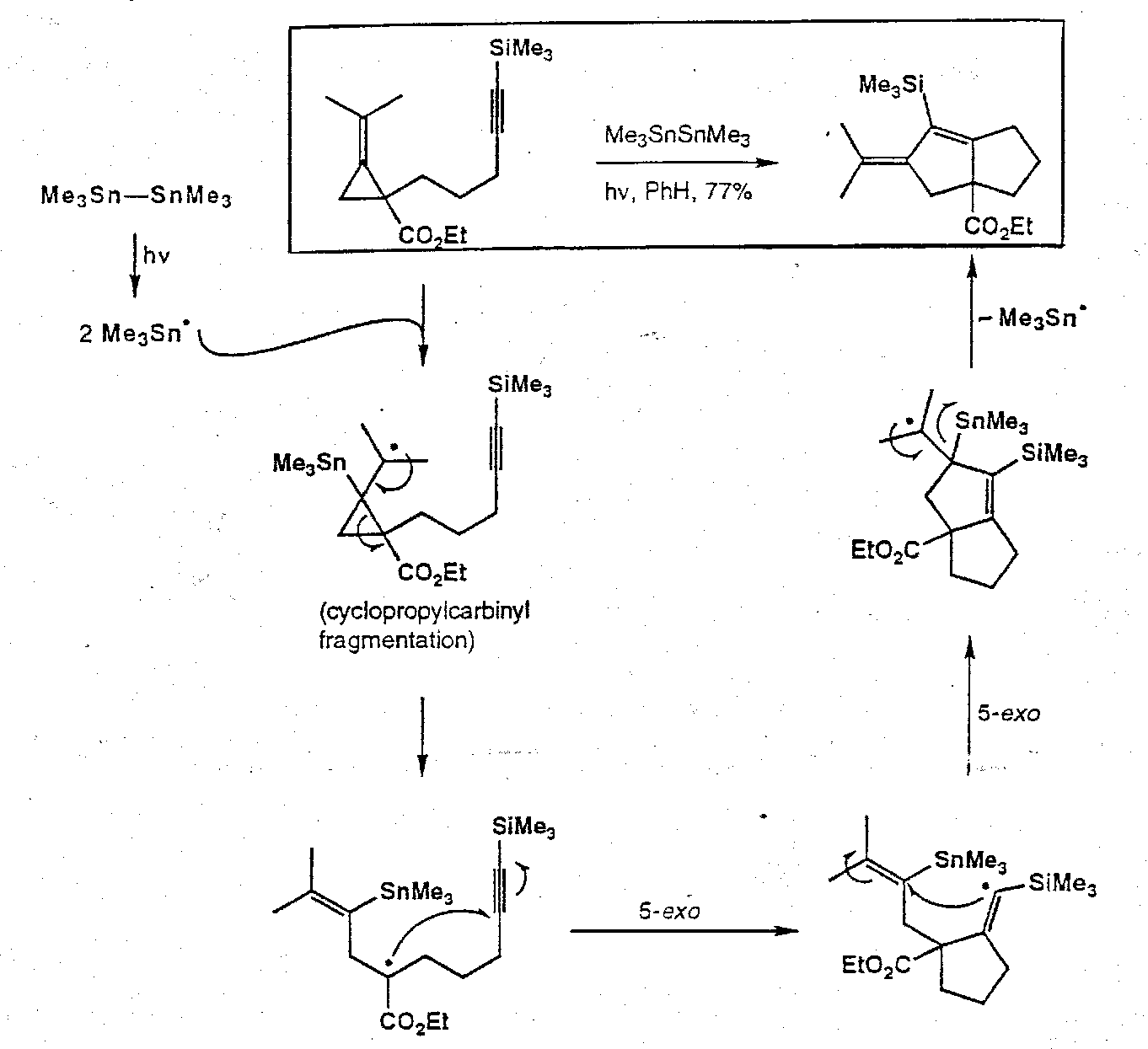
Cyclopropylcarbinyl Rearrangement
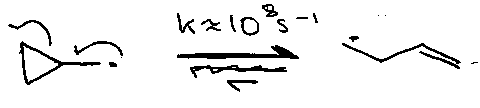
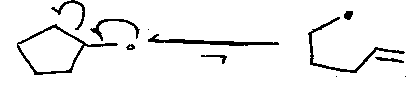
But,
Rate of H-atom Transfer –
R• + H-SnBu3 → RH + •SnBu3 k = 2.5 x 106 M-1 s-1
R• + H-SPh → RH + •SPh k = 1 x 108 M-1 s-1
Reason:
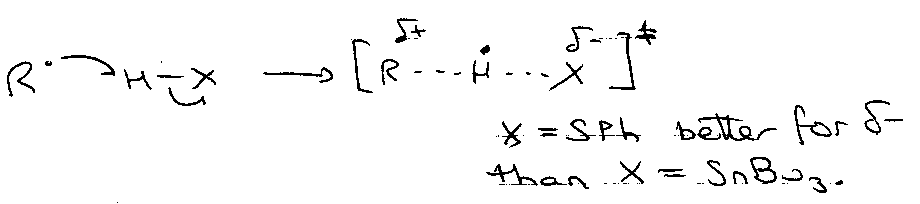
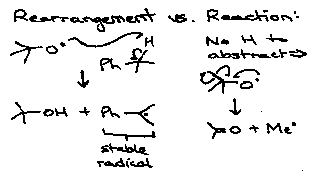
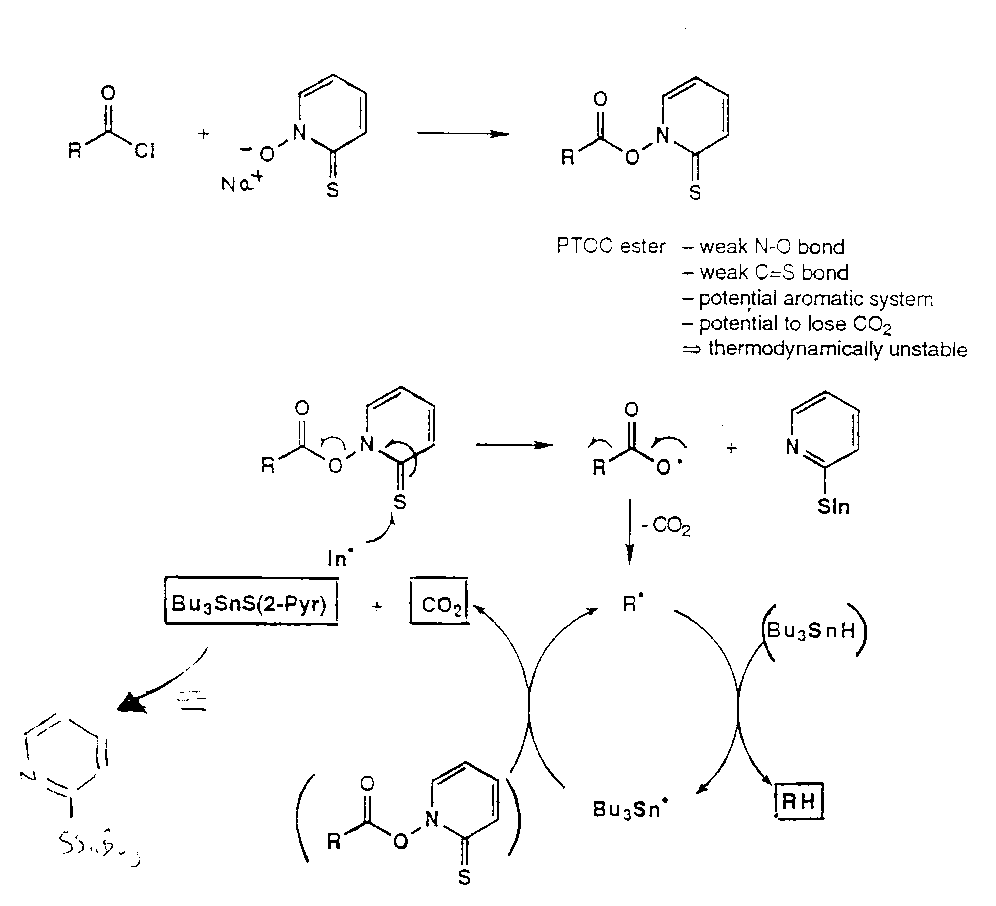
Barton’s Pyridinethione Oxycarbonyl Esters (PTOC esters)
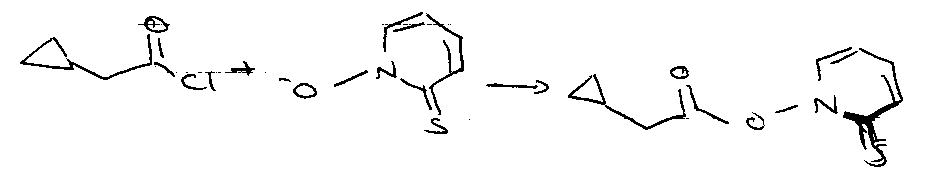
Consider:
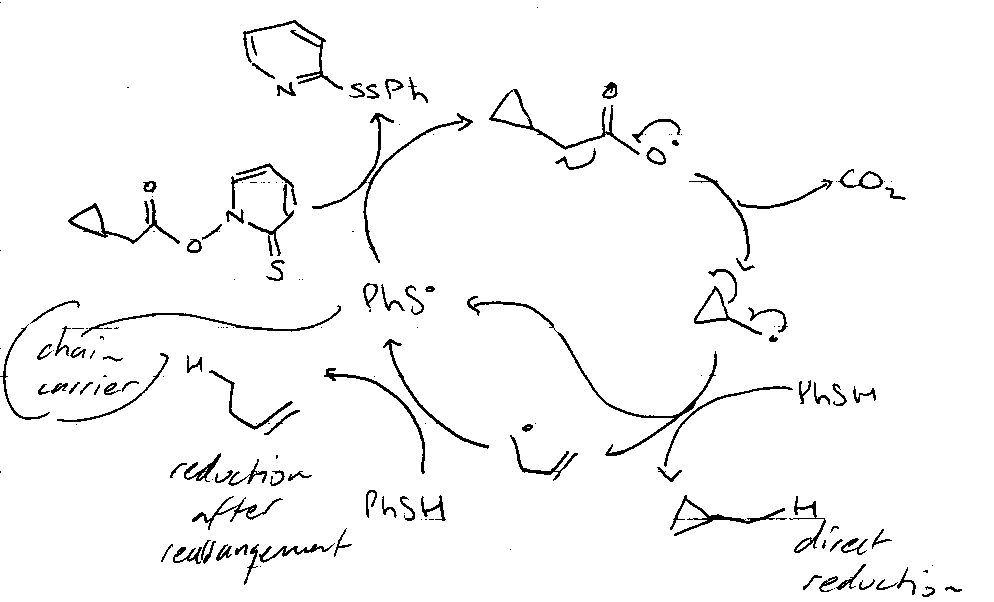
Via:
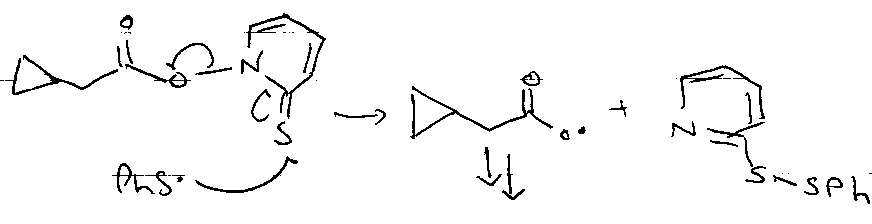
- C=S weak π-bond.
- PhS• attacks S=C → S-S bond.
- Aromaticity driven.
Radical Clocks
Free radical reactions having a known rate against which other reactions may be gauged (common is cyclopropylcarbinyl cleavage).
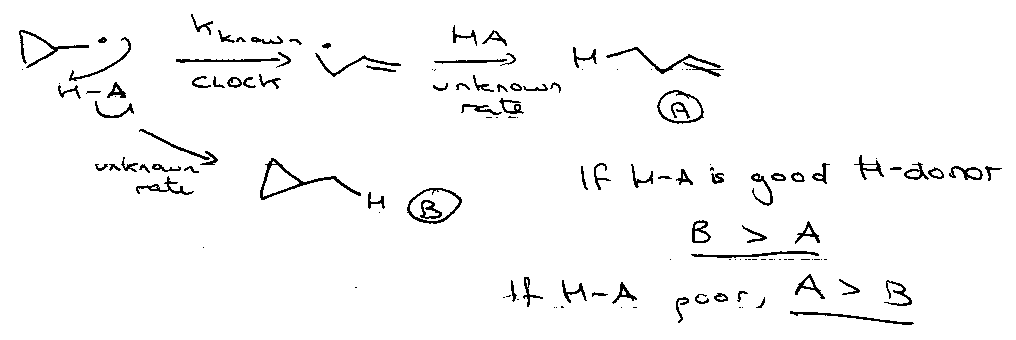
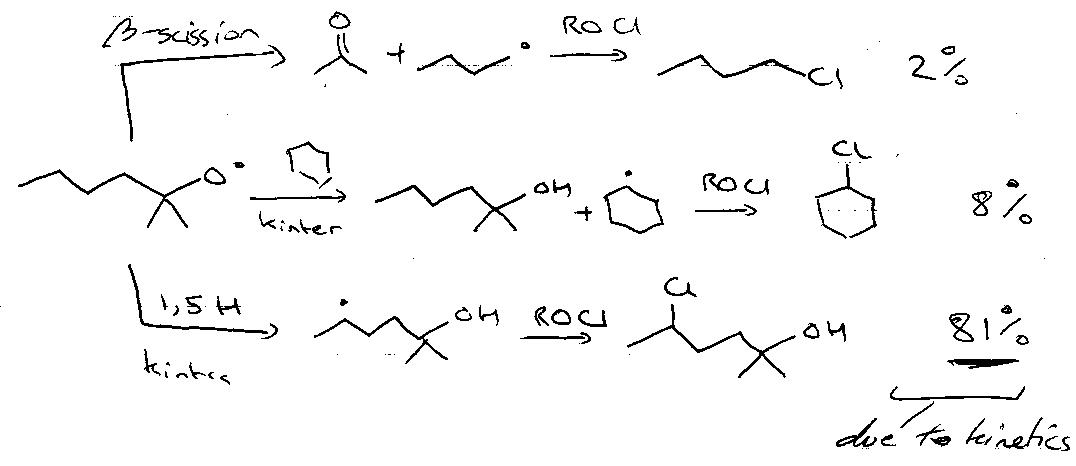
β-scission
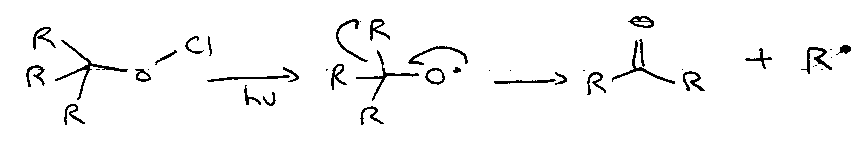
Effect of varying attached groups:

R = Ph → Me• ejected, as:
1) Phenyl destabilised wrt methyl, and
2) Favourable phenyl-carbonyl π-overlap may develop in the transition state.
Cyclisation
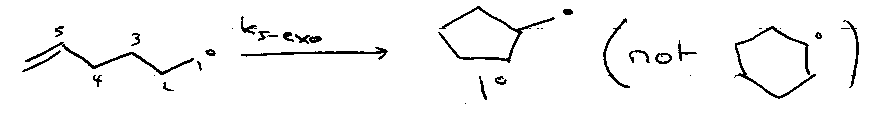
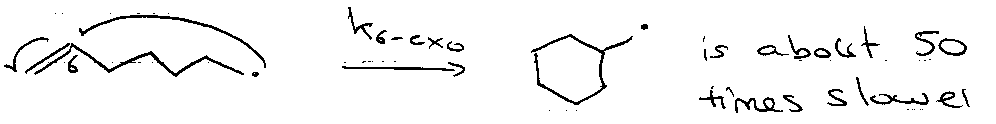
Follows kinetic control:
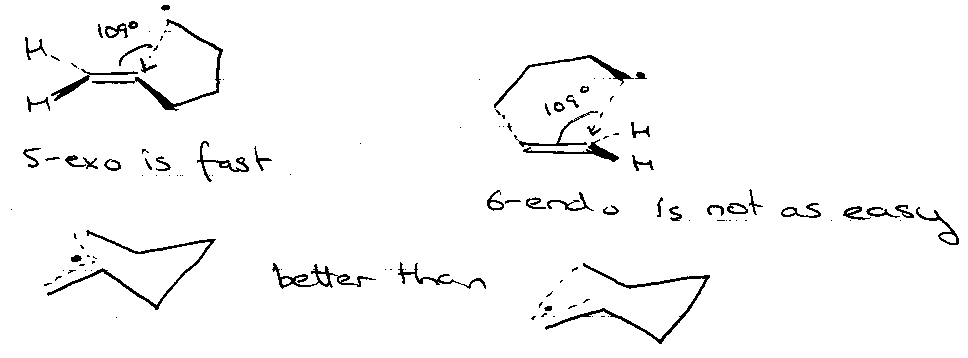
Baldwin’s Ring Closure:
Most important here are the trigonal systems –
3 to 7-exo-trig FAV
3 to 5-endo-trig DISFAV
6 to 7-endo-trig FAV
But thermodynamic control also? (Julia)
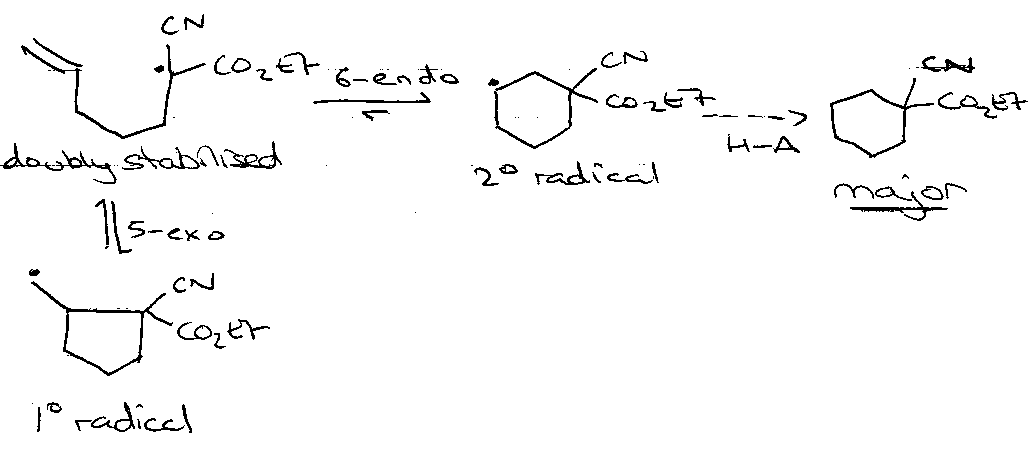
Balance of:
- Radical stabilisation, and
- σ vs. π C-C bond strength
5-exo-cyclisation followed by cyclopropylcarbinyl fragmentation also gives 6-membered rings:
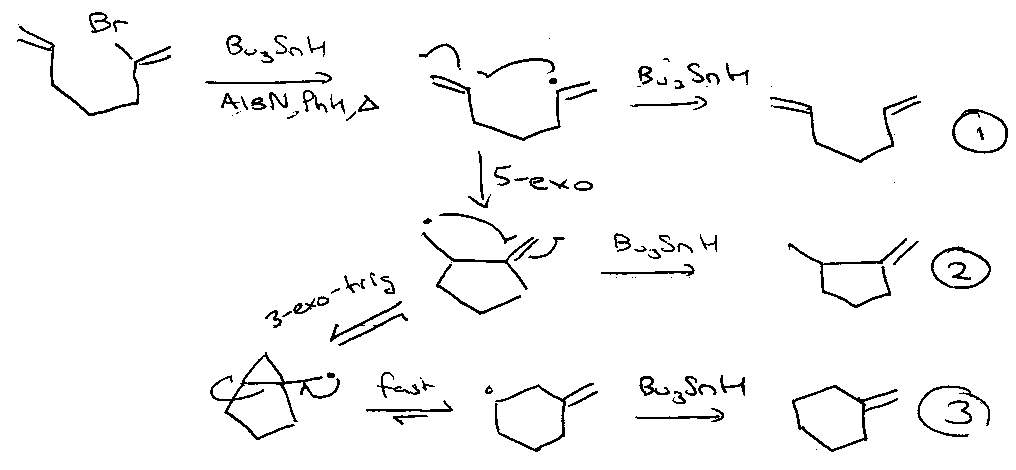
Depends on [Bu3SnH] – favours (1) >> (2) > (3).
5-exo-cyclisation as a mechanistic probe –
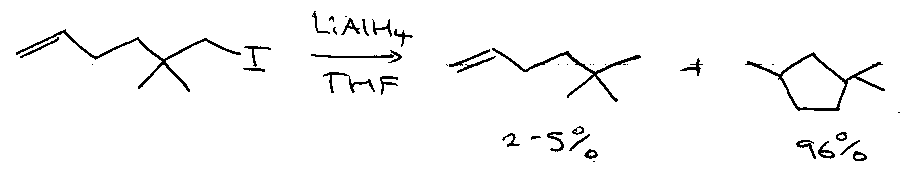
Ashby proposed:
SET by AlH4-
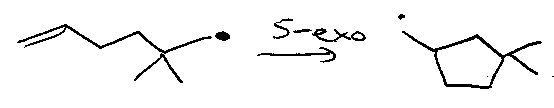
(Probably correct).
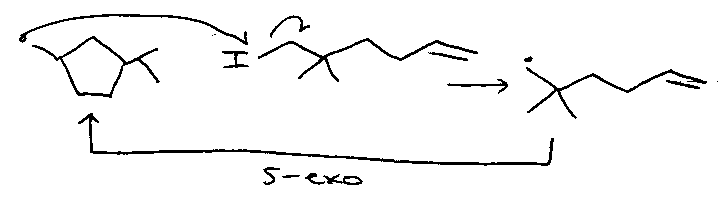
Newcomb proposed:
Iodine atom transfer / reduction sequence, via:
Radicals in Synthesis
Functional Group Chemistry
General Points –
- C-centred radicals are extremely reactive, yet they can be generated under mild, neutral conditions and often undergo highly regio- and stereoselective reactions.
- Radical additions to C=C are usually exothermic and irreversible with early, reactant-like Transition States. Kinetically controlled.
- Since radicals are not cluttered with counterions or solvation spheres, radical intermediates are ideally suited for synthesis at crowded bonds.
- C-centred radicals are inert to OH and NH, therefore no protecting groups for these. Exception: phenols (capto stabilised).
- Unlike carbanions, carbon radicals are not subject to β-elimination of OR or NR2.
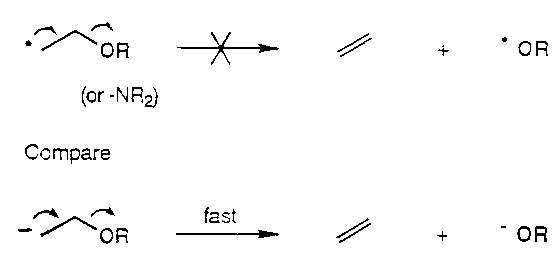
- Unlike carbocations, carbon radicals are not subject to capture by β-OR or –NR2 groups, nor are they usually prone to migration of β-H or –CR3 groups. They are, however, subject to β-elimination of SR, SOnR and SnR3 groups.

- Radical centres do not usually retain stereochemistry. Can be a drawback, but precursor synthesis is simplified (geometrically labile sp2-like radicals).
Examples
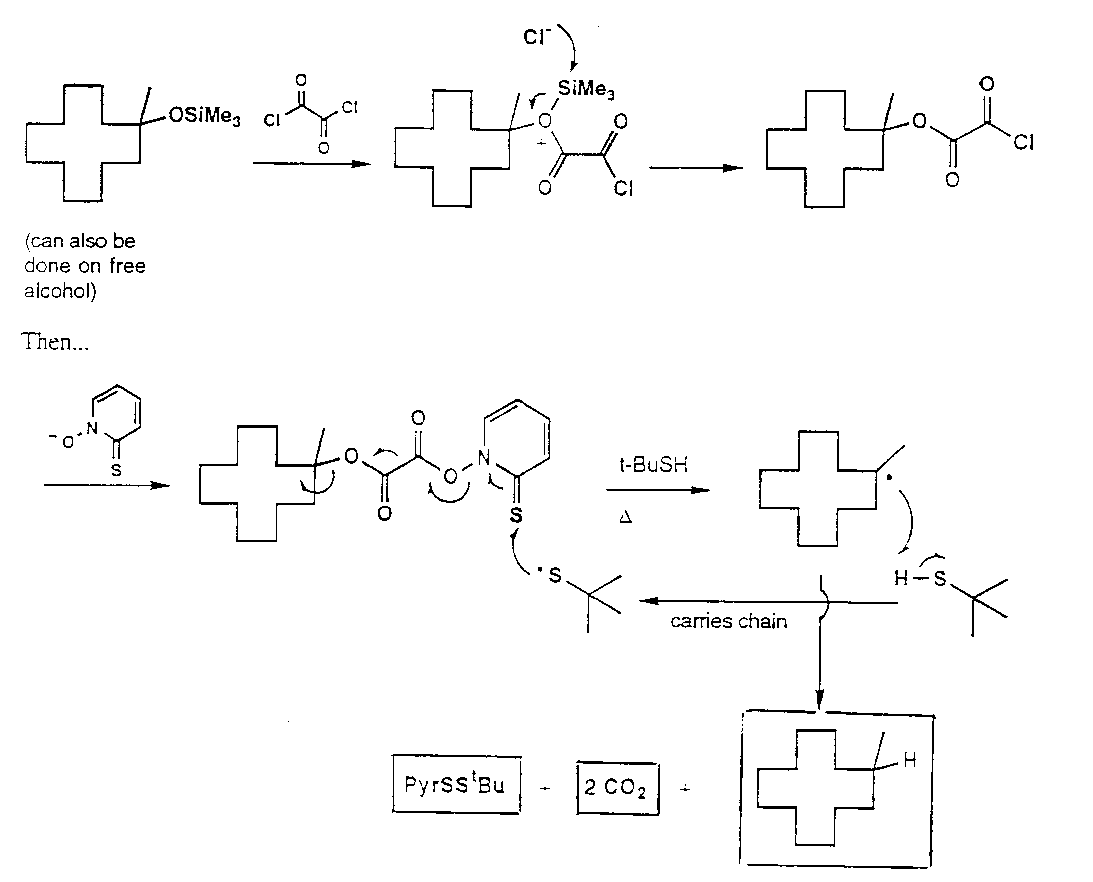
Barton Nitrite Ester Reaction
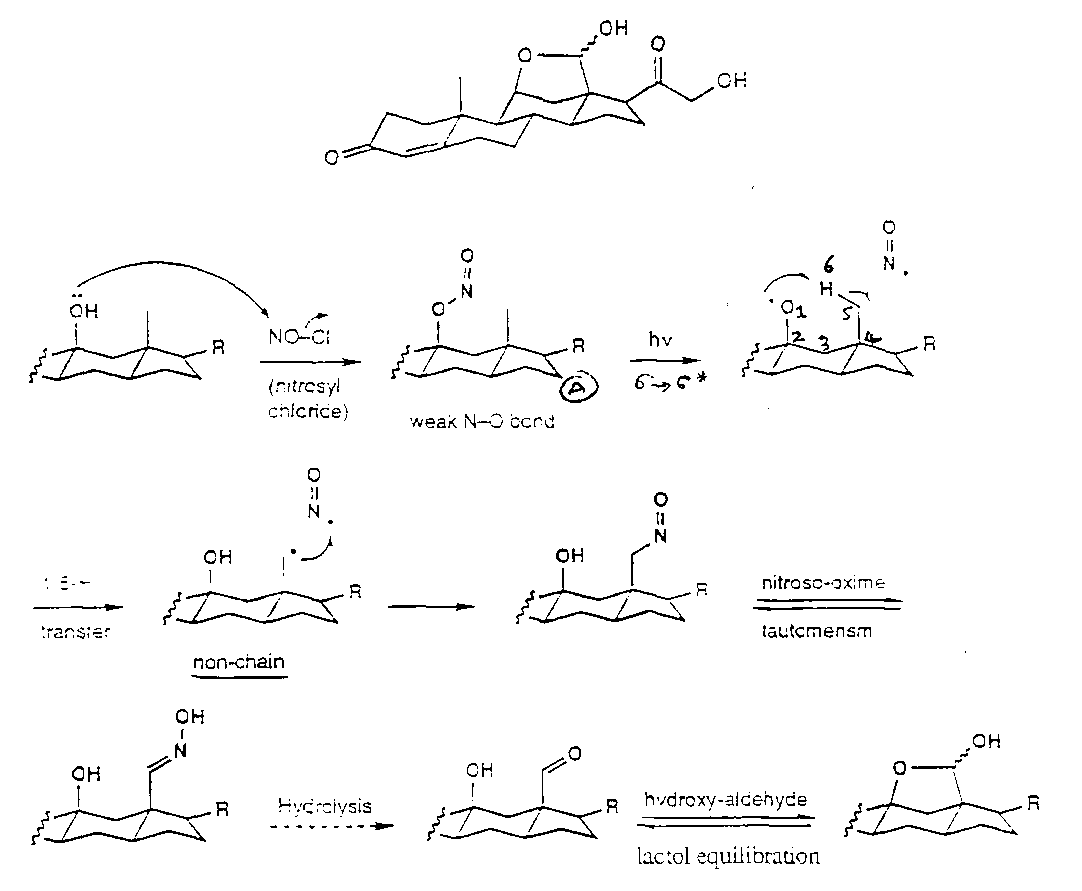
Hoffmann-Loffler-Freytag Reaction

Barton’s PTOC Esters
If there’s no added reducing agent (e.g. Bu3SnH):
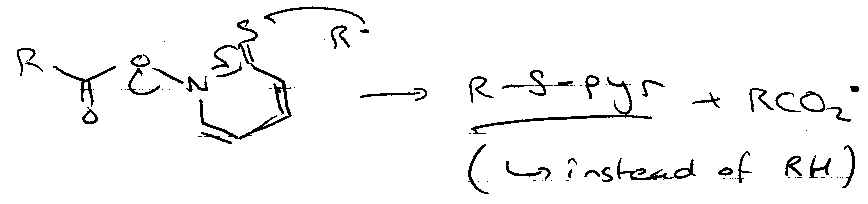
This leads to many possibilities for reaction:
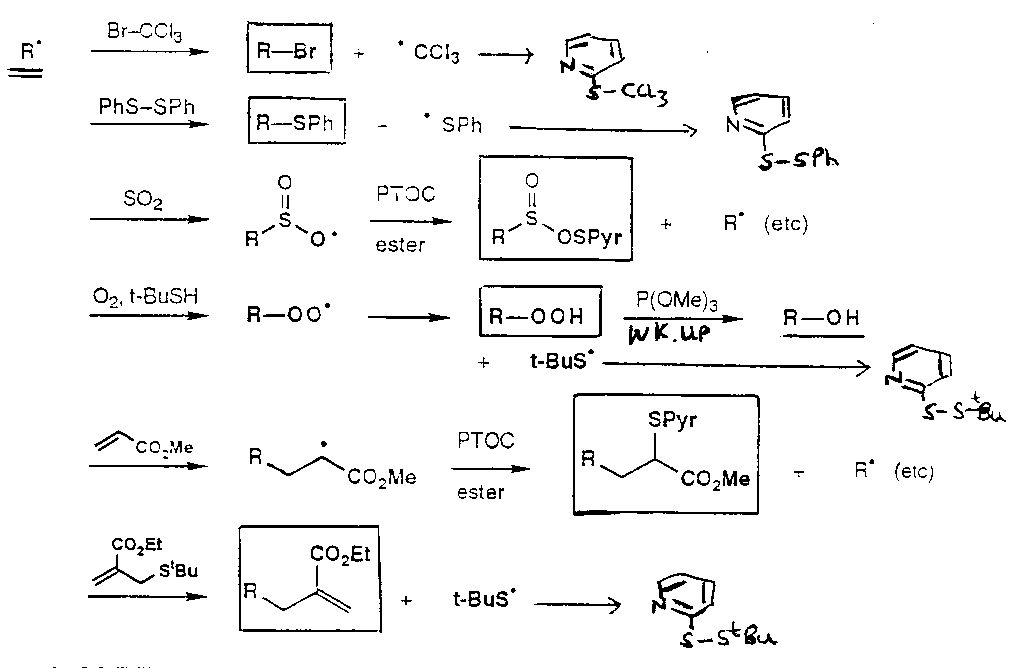
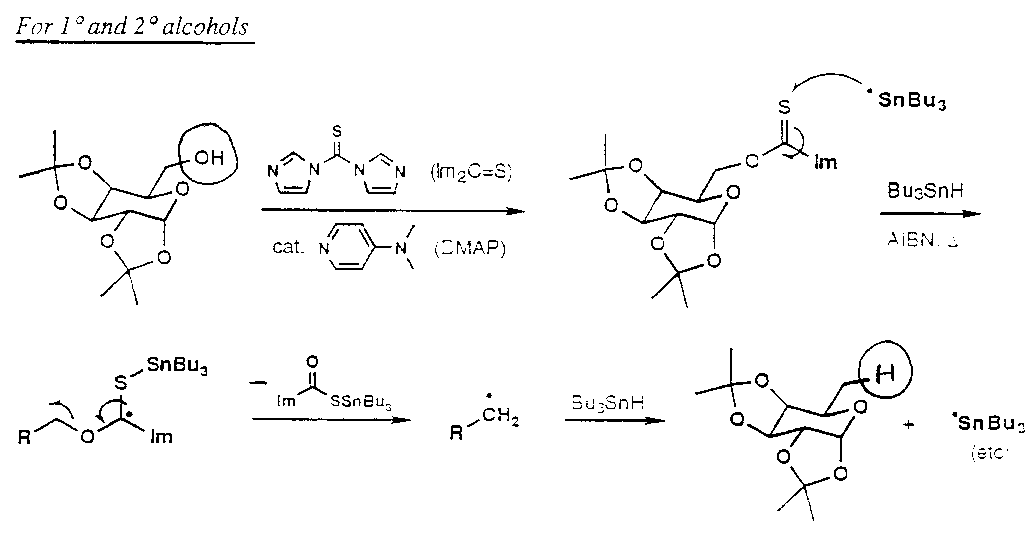
Removal of OH
Free Radicals in Natural Product Synthesis
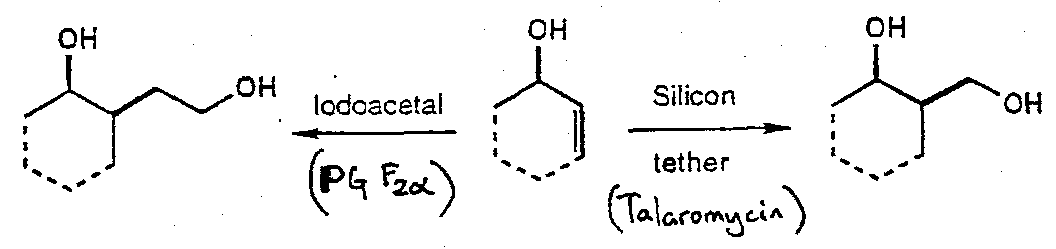
Prostaglandin F2α
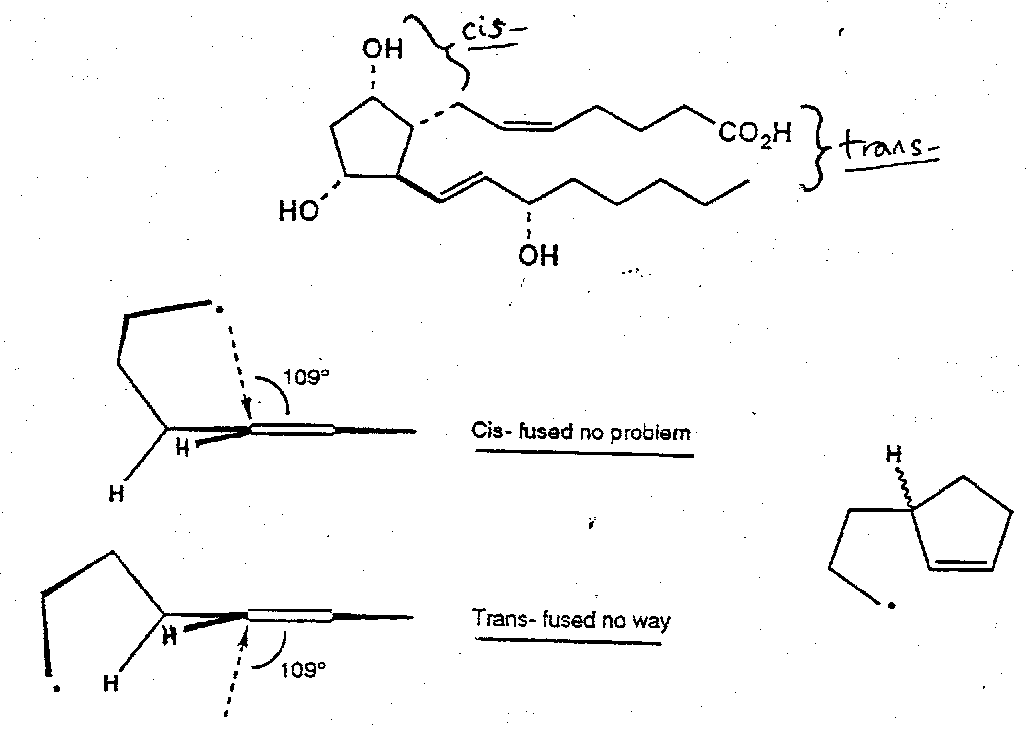
Iodoacetal tethered cyclisation –
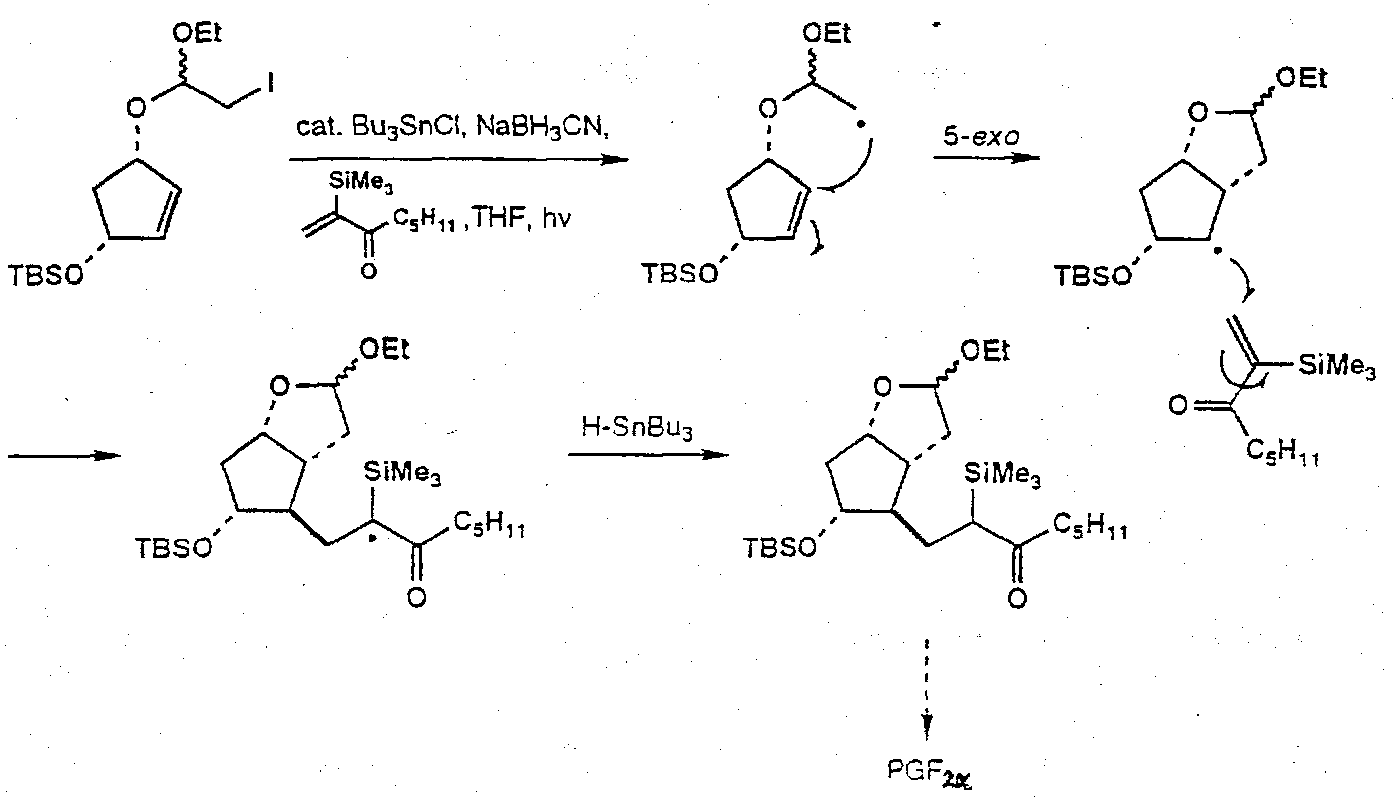
Notes:
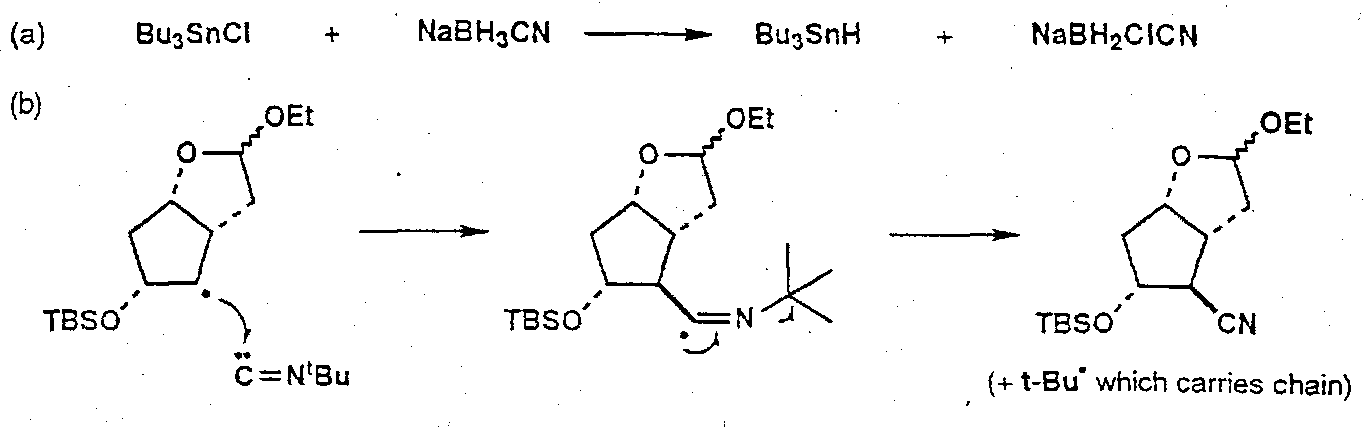
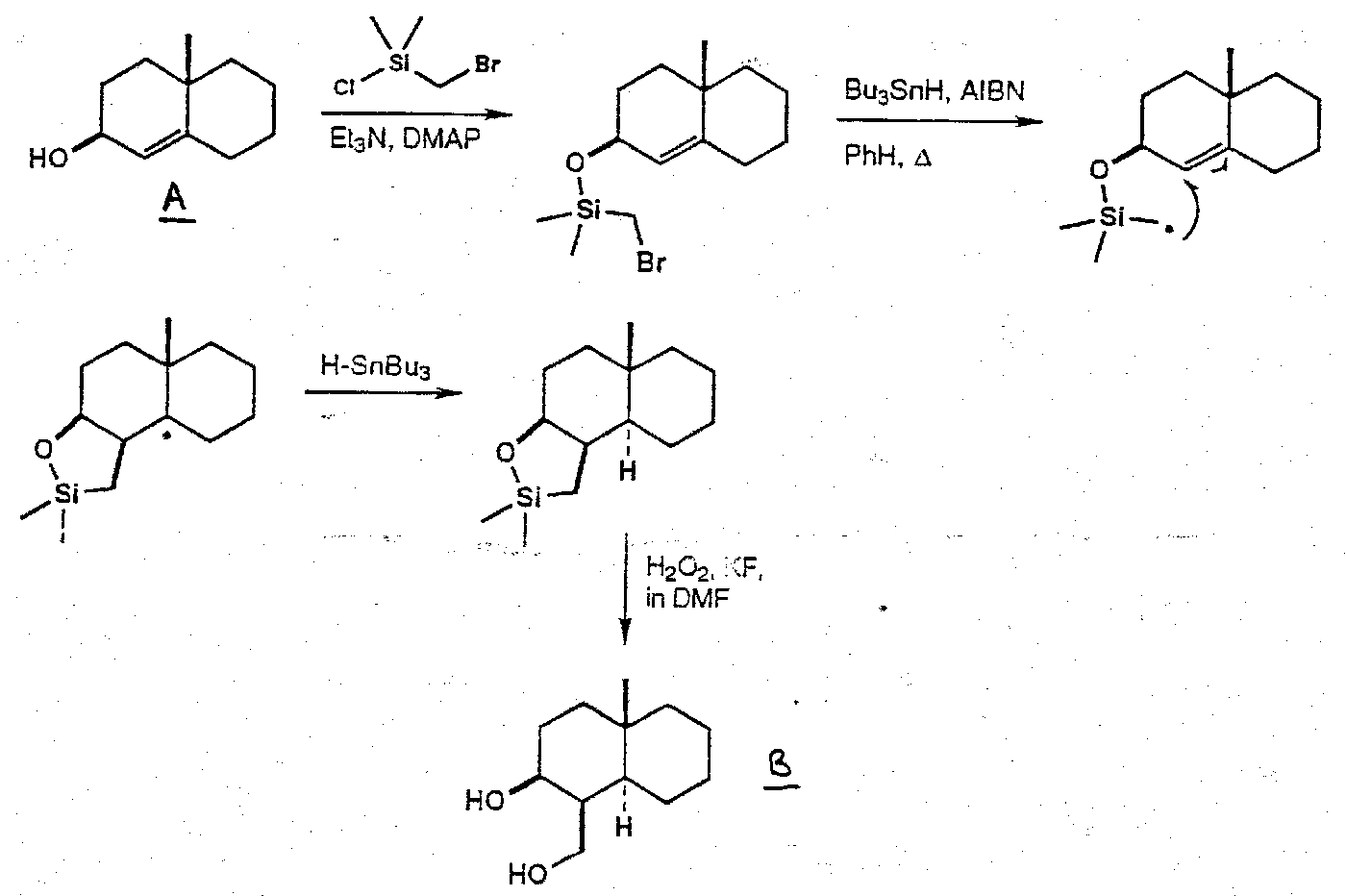
Also, Silicon Tethered free radical reactions –
Talaromycin A

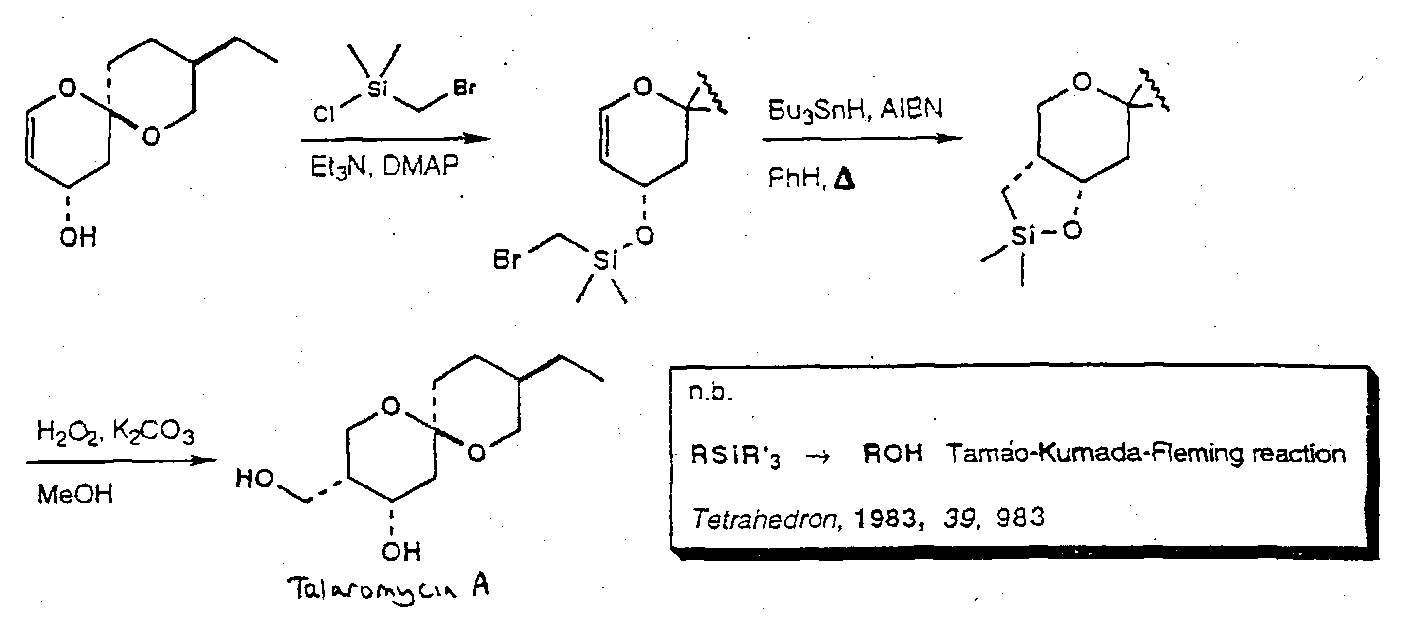
Comparison of iodoacetal and silicon tethered radical reactions
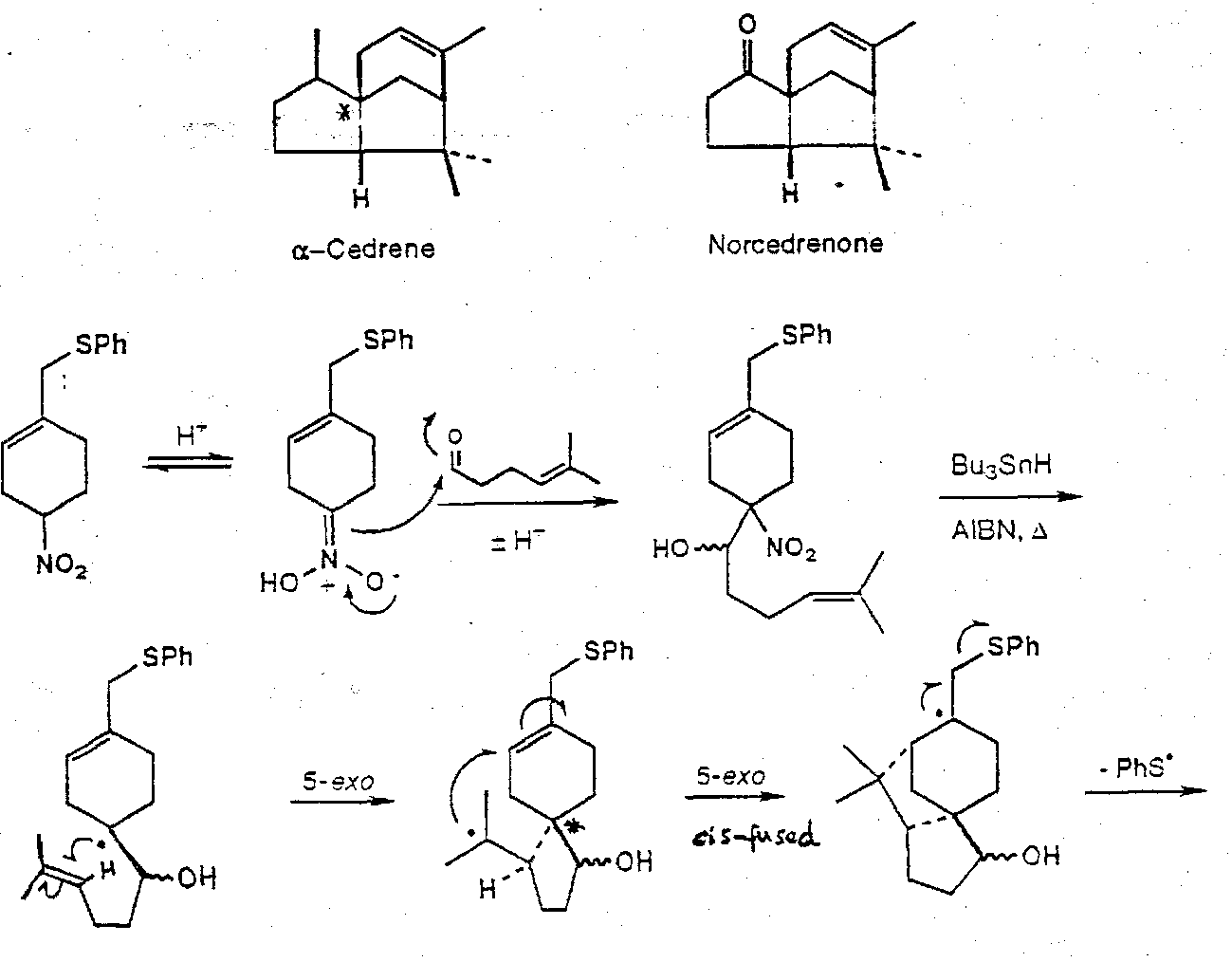
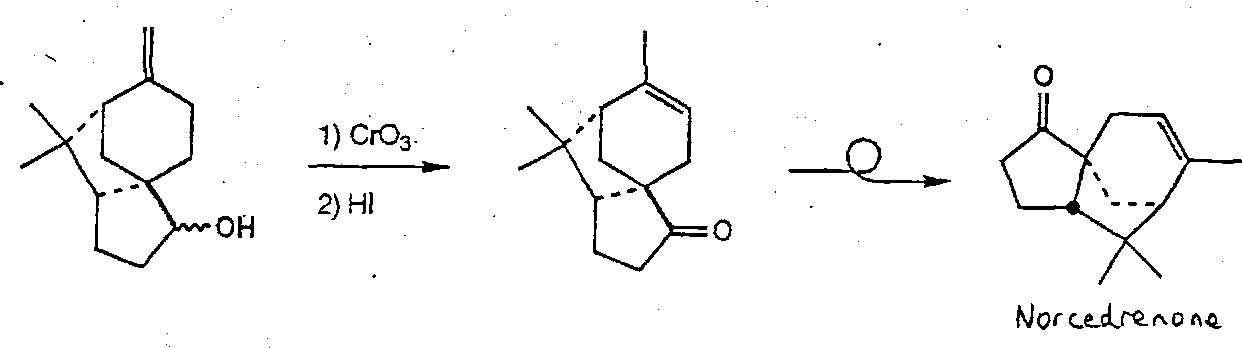
α-Cedrene
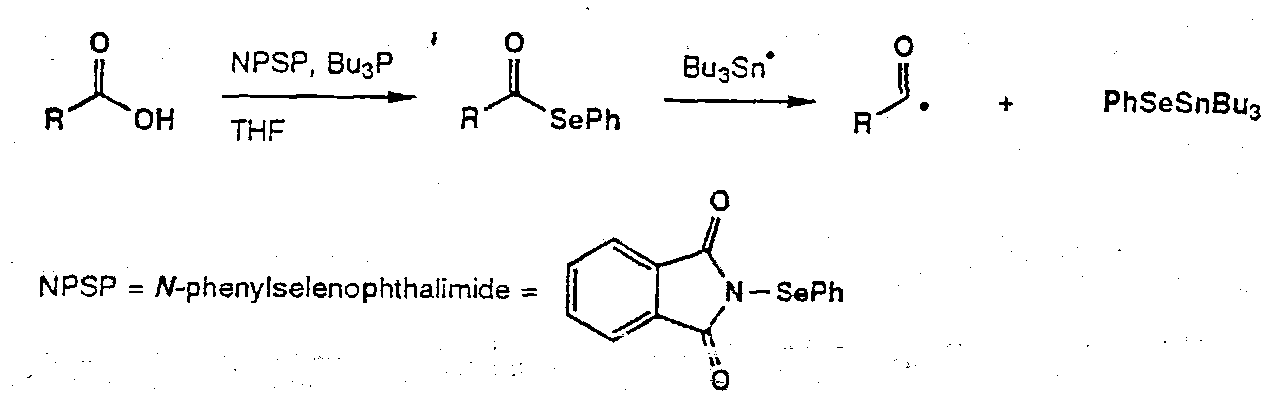
Acyl Selenides as acyl radical precursors
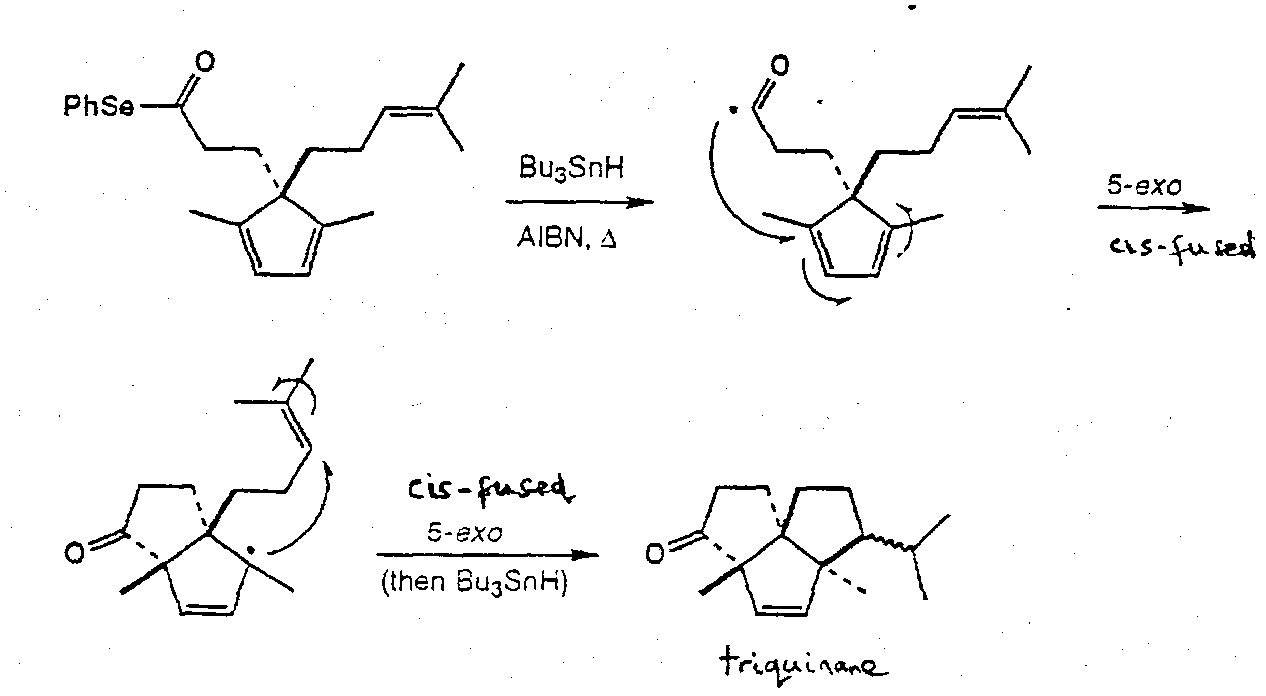
Fragmentation Chemistry in Synthesis
Intramolecular Annulation
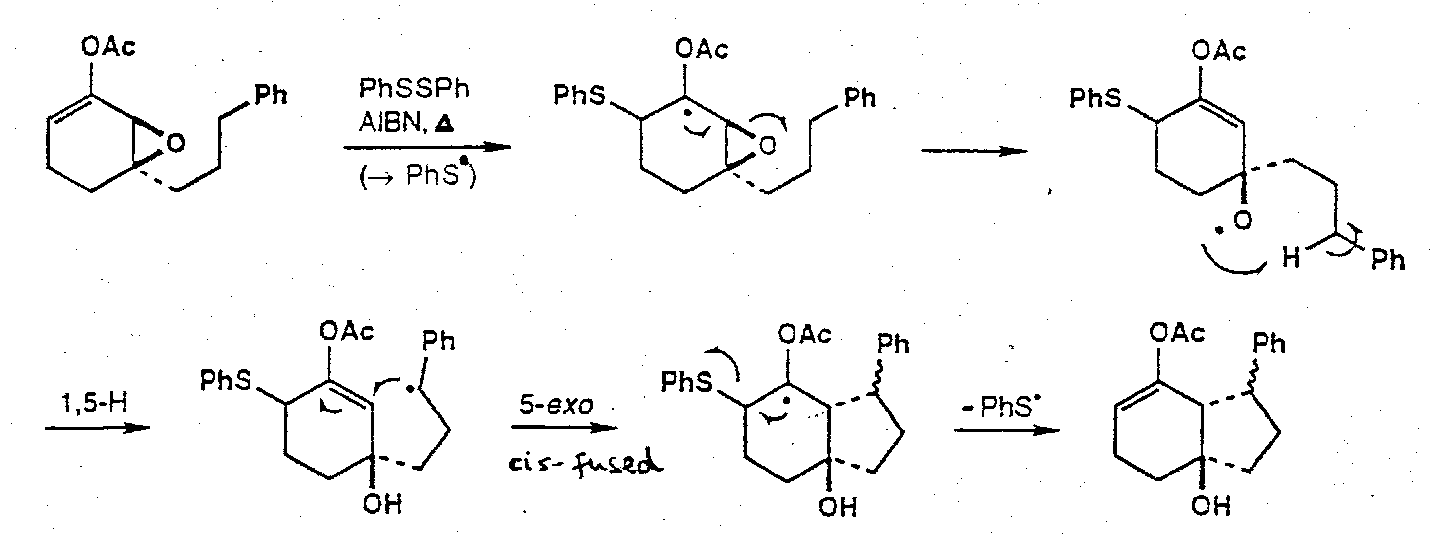
Epoxide Fragmentations
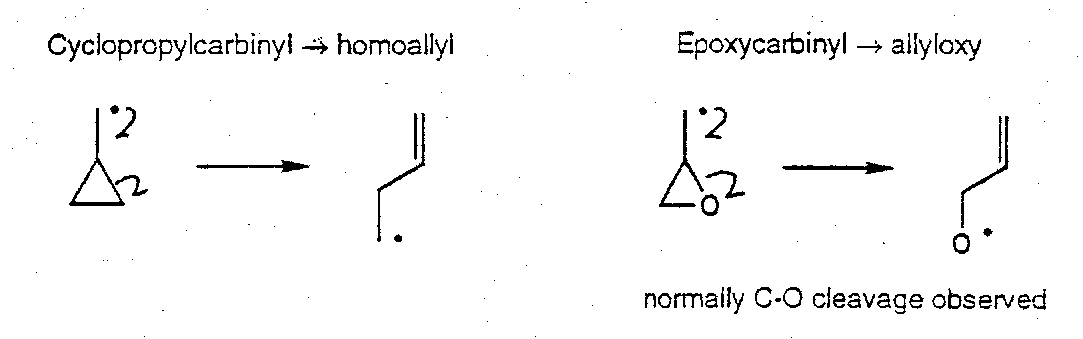
Incorporation into cascade sequences
I am happy for them to be reproduced, but please include credit to this website if you're putting them somewhere public please!