Descriptive p-Block Chemistry
Covers some of the descriptive chemistry of the p-Block, done by Group (12 to 18)
Descriptive P-Block Notes
GROUP 12
Zinc, Cadmium and Mercury –
These are being included in this p-block description because the full d-orbitals mean their properties are more p-block in character than Transition Metal.
Occurrence –
Chalcophiles. Major ores are ZnS, CdS and HgS (only source of Hg). Extraction usually involves roasting to oxide then reduction over C, although HgS + O2 gives Hg + SO2 directly.
Elemental Form & Allotropes –
They have noticeably lower melting points and boiling points. Mercury is a liquid at room temperature. Hg is also unique in that it is the only element apart from the noble gases that is a monatomic vapour. Zn and Cd are silvery solid with bluish lustre. Liquid Hg has exceptionally high electrical resistivity for a metal.
Zn and Cd show distortion of metallic HCP. All these metals have widely separated coplanar atoms giving lower density and tensile strength than Group 11. This is due to the stability of the d electrons now being tightly bound to the nucleus, such that metallic bonding uses only s electrons (weak).
Reactivity –
Zn and Cd tarnish in air and will combine with O, S, P & X on heating. Mercury also reacts with these (except P). Non-oxidising acids dissolve Zn and Cd, but not Hg. Zn is the only element which dissolves in aqueous alkali. All 3 elements are often used in alloys, e.g. brass and amalgams of mercury. The trend is for Zn and Cd to be very similar in reactivity, while Hg is quite different.
Zn and Cd are more electropositive, while Hg is comparatively inert. With the exception of metallic radii, it appears that the effects of Lanthanide Contraction have ceased by Group 12.
The d10 configuration leads to almost exclusively MII chemistry. This is shown by the ionisation energies, where s electrons are easily removed but d are firmly held. EI is still high for mercury, due to poor nuclear shielding from the filled 4f. Hence there is a positive electrode potential for Hg couple.
Zn is similar to Mg (class-a, therefore donor ligands). However Zn has a greater tendency to form covalent compounds as well, hence complexes with S and N ligands, and also X and CN-. Cd is similar to Zn, but falls on the class a/b borderline. Mercury is definitely a classic case of class-b, forming very stable complexes with N,P & S donor ligands.
Polarising Power MgII < ZnII < CdII < HgII leads to colour changes due to charge transfer from ligands. This trend is a reflection of decreased nuclear shielding and increased power of distortion in the sequence: filled p < filled d < filled f. This trend also leads to increased stability of σ-bonded alkyls and aryls down the group. Hg-C bond is not strong, but the competing Hg-O bond is weaker. Thus mercury has many stable organometallic compounds.
The MII ions do not form π-complexes with CO etc, due to stability of d10 – doesn’t want to provide electrons for back-bonding. Also the filled d prevents π-acceptance.
Types of Compounds –
Carbides and Nitrides of these compounds are unstable (Hg ones explosively so). Of the hydrides, ZnH2 is a moderately stable white solid. CdH2 and HgH2 are much less stable and decompose rapidly even below 0oC.
Oxides and Chalcogenides
Mostly zinc blende or wurtzite structure reflects preference for tetrahedral coordination. The normal oxide is MO, although the peroxide MO2 is known for Zn and Cd. Lower oxides involve mixtures with elemental metal.
ZnO is formed by burning Zn. Its colour changes to yellow on heating due to evaporation of O2 leading to a nonstoichiometric phase. It is actually possible to form a whole range of colours. ZnO is amphoteric.
CdO lattice defects lead to a wide range of colours. It is more basic than ZnO (dissolves readily in acid and hardly at all in alkali). HgO can be red or yellow.
ZnS as zinc blende is more widespread but wurtzite is more stable at high temperatures. Pure ZnS is white, and it dissolves readily in mineral acids releasing H2S, but roasting renders it much less reactive.
Cd chalcogenides are similar to those of Zn and display same duality of structure. Many stable pigments of brilliant colours. HgS is polymorphic and unreactive, requiring conc. strong acid to dissolve.
Halides
All 12 dihalides are known, and 4 other halides of Hg22+ form.
The difluorides are very distinct from the other dihalides, e.g. much higher mp/bp, which suggests they are ionic. Other halides of Zn and Cd are hygroscopic and very soluble, leading to hydrates. Significant covalent character is indicated here by low mp, solubility in organic solvents and layered lattice (2D) structures. Conc. aq. ZnCl2 dissolves starch, cellulose and silk, therefore cannot be filtered through paper. It is useful in textiles industry.
As expected, the mercury halides show more covalency. They are low mp volatile solids, soluble in most organic solvents. Solubility in water decreases with weight, as they exist as HgX2 molecules.
Hg2X2 have low solubility in water, and are easily volatilised. Their vapour density would indicate HgX, but the diamagnetic nature indicates that it is in fact Hg + HgX2.
Mercury(I) Compounds
These are known. Note that they are diamagnetic, whereas Hg+ would be paramagnetic.
Polycations Hg22+ form by overlap of 6s orbitals, with little involvement of 6p or 5d. They can coordinate another ligand, which means Hg catenation could be expected. In fact, Hg32+ is linear, and Hg42+ also exists and is almost linear (Oxidation State is equivalent to +½). This can be formed using SbF5 or AsF5 (adducts).
Zinc(II) and Cadmium(II) Compounds
Salts of most anions known, but invariable oxidation state of +2. Oxosalts are often isomorphous with MgII, but have lower thermal stability.
Carbonates, Nitrates and Sulphates all decompose to oxide on heating. Aqueous solutions are appreciably hydrolysed to [M(OH)(H2O)x]+. There is some coordination chemistry, but it is less extensive than other transition metals. No stable fluoro complexes, but [MX3]- and [MX4]2- are moderately stable. Tends to favour tetrahedral coordination number, and those higher than 6 are very rare.
Mercury(II) Compounds
Only ionic compound is HgF2. Any compound with appreciable charge separation, e.g. strong oxoacids, are extensively hydrolysed. Their ionic character is symptomatic of the reluctance of Hg(II) to form covalent bonds to O.
Hg has a characteristic ability to form not only ammine and amine complexes, but direct covalent bonds to N. Also it can form cluster compounds due to Hg-M bonds. There are also a vast range of stable organomercury compounds.
GROUP 13
Boron –
Occurrence –
Occurs as borate minerals and borosilicates.
Can be obtained in elemental state by reducing B2O3 with Magnesium, although there are several other methods.
Elemental Form & Allotropes –
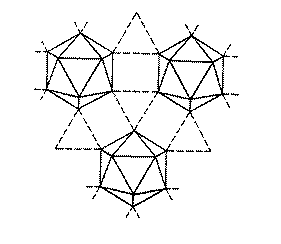
Dominant allotrope is an icosahedral (B12) unit. This is very open structure (only 37% space filling). The 36 valence electrons divide into 26 for the bonds in an icosahedron, then 6 more for standard 2 centre 2 electron (2c2e) bonds, and the remaining 4 form 3 centre 2 electron (3c2e) bonds. This is an illustration of a common occurrence in boron chemistry – multi-centred bonding.
There are however an extremely large number of possible allotropes. This is due to boron seeking to solve the problem of having fewer electrons than orbitals available for bonding, coupled with the fact that it cannot form metallic bonds due to its small size and high ionisation energy (covalent only).
Reactivity –
Unlikely other Group 13 Elements – higher ionisation energy and non-metallic. One less electron than number of valence orbitals means it is essentially “electron deficient”. This means it is often a Lewis Acid (accepting electron pairs) and forms multi-centred bonds.
It also has a high affinity for Oxygen, and its small size allows it to occupy interstitial sites in metal borides.
Boron itself is quite inert at low temperatures (though it will react with F2 readily), but at higher temperatures will react with almost all metals and non-metals.
Range of Oxidation States –
Electron configuration is [He]2s22p1, so the main ox. state is +3. The ionisation energy is very high and B3+ is very small, which means that the ionic form is highly unstable.
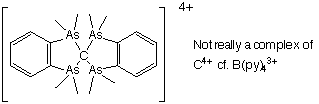
Thus, B3+(F-)3 will readily form BF3, and so there is no cationic chemistry, except for one major example:
BBr3 + 4 (py) → [B(py)4]3+(Br-)3
Since there is no cationic chemistry, there is also no redox chemistry in solution.
Types of Compounds –
There are 5 major types of boron compounds:
- Metal Borides
- Boron hydrides and their derivatives.
- Boron halides
- Oxo compounds (borates, silicates).
- Organoboron compounds + B-N compounds.
Borides
There is an enormous range of metal boride stoichiometries possible, as well as non-stoichiometric phases with variable composition. Chains, layers and polyborides form, such as B6 units. They are typically used in armour and rocket nozzles (inert).
They can be prepared in a variety of ways to suit, the simplest being direct combination of metal and elemental boron at high temperature. However, in most cases it is very hard to acquire pure products of the precise stoichiometry.
The most common stoichiometries (75% of them) are M2B, MB, MB2, MB4 and MB6. Transition Metals tend to favour metal-rich borides while Groups 1-3, and the lanthanides and actinides tend to form Boron-rich borides.
Boron Hydrides
Many possible boranes can be formed.
BF3/NaBH4 is best method for producing B2H6.
The reaction with MgB2 and water gives hexaborane-10.
They are all endothermic compounds (thermally unstable due to strong B-B and H-H bonds in elemental states). They are also colourless and diamagnetic. They are kinetically reactive but reactions do not proceed due to a weak B-H bond (it is actually strong) but instead due to availability of alternative structures and vacant orbitals of similar energies.
Converting to higher boranes is achieved by controlled pyrolysis conditions.

Oxidation states of less than 3 indicate B-B bonds present. They are reducing agents (but not in aqueous). B-H → B-O is favoured since the hydride ligand is transferred. Studies of boranes illustrated the 3c2e bonding (B-H-B) required bridging H atoms.
BH3 exists only in very small concentrations when reactions are taking place (as shown by matrix isolation techniques). It can be generated by thermal dissociation of diborane using Lewis Bases (to give L.BH3). The relative stabilities of L.BH3 show the following series:
L = PF3 < CO < ethers < organic sulphides < py < H-.
The higher presence of sulphides than ethers shows that BH3 shows some class b acceptor (“soft acid”) characteristics despite the absence of low-lying d-orbitals on boron.
In addition to pyrolysis and cleavage, Diborane also undergoes substitution and solvolytic reactions. For example, reacting it with HCl yields B2H5Cl, while reacting with Cl2 gives BCl3. It also undergoes hydrolysis to give B(OH)3 and hence is acidic:
B2H6 + 6H2O → 2B(OH)3 + 6H2 → B(OH)4- + H3O+
MBH4 compounds are formed by reaction of B2H6 with MH compounds, and the resulting reagent is very useful for reduction of organic compounds.
Boron Halides
Trihalides can be synthesised as follows:
B4O72- + “HF” → 7H2O + 4BF3.
B2O3 + 3Cl2 + 3C → 2BCl3 + 3CO (dry)
NaBH4 + 2I2 → BI3 + NaI + 2H2
They are planar, monomeric, strong Lewis acids (complex), and resemble Organoboranes in this respect. Their Acidity gets stronger down group – this is against the electronegativity trend, and is explained by π-bonding (resonance across all 3 bonds) giving a strong intramolecular bonding monomer. The empty B orbital gets filled easily.
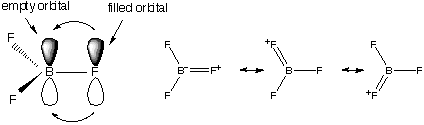
Compare with Al:
Al2Cl6 => weak intramolecular π-bonding dimer
BCl3 => strong intramolecular π-bonding monomer
They react with water to yield HX + H3BO3, except when X = F, where a H3O+[BF3(OH)]- complex forms which reforms BF3 (B-F is very strong).
Lower halides (B2X4 etc) all have a long B-B bond, and X = F is planar (D2h). X = Cl, Br, I have a tendency to form a staggered D2d structure.

They are isoelectronic with N2O4. They are formed most commonly by an electric discharge over mercury or copper electrodes, and [BX] inserts into BX3 directly.
They are not very stable, and spontaneously flammable in air and disproportionate readily.
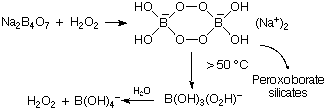
Boron Oxides
Forms B2O3, which tends to B(OH)3 in water, which is acidic. It is a weak acid since it acts solely by –OH accepting, and not by proton donation.
2B + 3/2O2 B2O3 B(OH)3
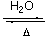
The structure is H-bonded, and planar BO3 have the π-bonding effect.
Boric acid is unusually still acid in H2SO4, forming B(HSO4)4-.
B(OH)3 + 2H2O B(OH)4- + H3O+
cf. HCl + H2O Cl- + H3O+
NB .
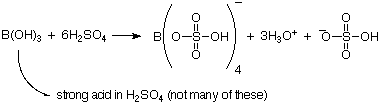
B2O3 + metal oxide → borates.
Borates are of the form [BxOy]n-. Boron can link either three or four oxygens. Polynuclear anions can be formed by corner-sharing only of boron-oxygen triangles/tetrahedral.
When hydrated, borates with protonate oxygen atoms in the order:
free O2- > tetrahedral O-B > triangular O-B > free –OH.
Polymerisation of borates occurs readily as water can be expelled.
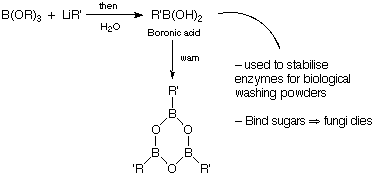
Boron-Nitrogen
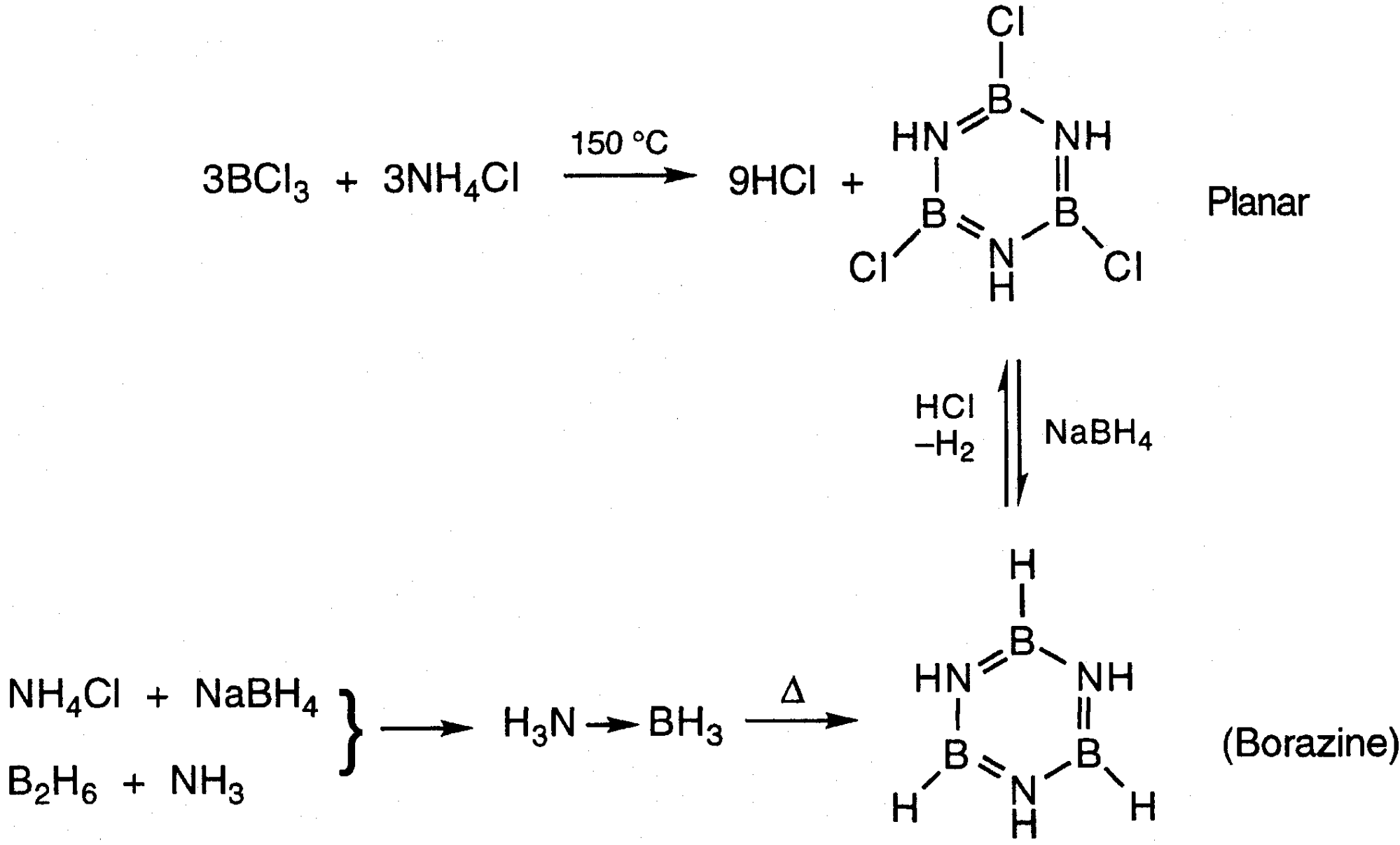
B=N is isoelectronic with C=C (just as B-N is isoelectronic with C-C), so borazine behaves like benzene, and is said to be aromatic as well. The ring is planar and all the bonds are the same length. It is, however, much more reactive than benzene due to its polarity of BN bond.
Borazine under heat will form boron nitride (BN)x.
hexagonal like graphite structure (layered)
cubic like diamond (hard)
Hexagonal (BN)x versus Graphite
Graphite: carbon in one layer on top of a ring (staggered)
Hexagonal (BN)x: B above N interlayer interaction (eclipsed)
Weak B-N interlayer interaction
(BN)x less easy to cleave.
polar B-N bond means that electrons are localised, and so (BN)x is an electrical insulator.
There is the possibility of much repetition of organic chemistry in this way by replacing C-C with B-N.
Other Group 13 – Aluminium, Gallium, Indium & Thallium –
Occurrence –
Aluminium is the most abundant metal in the earth’s crust and present in many minerals (often oxides and silicates). It is extracted from bauxite by first isolating Al2O3 and then using electrolysis.
Ga, In & Tl are far less abundant, and tend to occur as sulphides rather than oxides.
Gallium is always found with Zn and Ge (its neighbours in the Periodic Table), or Al. It is usually extracted as an impurity from bauxite.
Indium and Thallium are recovered from the roasting of sulphide ores of Zn/Pb as by-products in H2SO4 manufacture.
Elemental Form & Allotropes –
They all differ greatly from Boron (a covalently bonded, non-metallic insulator).
They are all low-melting point, soft metals with low electrical resistivity. Heats of fusion and vaporisation also low, and tend to decrease with increasing atomic number.
There properties are very similar to the neighbouring metals on either side – this is due to a similar number of electrons being available for metallic bonding.
|
Element |
Structure |
|
Al |
FCC |
|
Ga |
Orthorhombic similar to iodine * |
|
In |
Distorted – face centred tetragonal |
|
Tl |
HCP |
* Ga tends to dimerise to Ga2 units (partial pair-wise interaction of the single p electron). This makes it structurally very similar to Mercury.
Interatomic distances in Ga are less than in Al emphasising the presence of a d-block contraction. Ga is also unusual in that it contracts on melting (this also occurs with Ge, its neighbour, and Sb, its diagonal neighbour).
Reactivity –
Changes in reactivity are due largely to the inner core properties as well as the ns2np1 valence electrons. This is because for Al (and B) it is simply the preceding noble gas core, for Ga and In it is the noble gas plus d10, and for Tl it is the noble gas plus 4f145d10.
The noble gas core change has an obvious effect in the trend of ionisation energies. There is an expected decrease from B to Al, but then an increase from Al to Ga due to d-block contraction in atomic size and higher effective nuclear charge as a result (the d electrons do not completely shield the extra positive charge). The same effect is observed from In to Tl due to lanthanide contraction. This is known to be due to the noble gas core as the same trend is not observed in Group 3 (Sc to La).
In terms of reactivity, they are more reactive than boron at moderate temperature, and they also behave as cations (unlike boron – strictly covalent). A consequence of this is a far smaller range of hydrides and cluster compounds (i.e. boranes and carboranes).
Aluminium:
Aluminium combines with most elements, and has a high affinity for oxygen (it is often used as a reducing agent to purify transition metals – thermite process).
Aluminium Hydroxide, Al(OH)3, is amphoteric, reacting either as:
ACID: [Al(H2O)3(OH)3] + 3H3O+ → [Al(H2O)6]3+ + 3H2O
BASE: [Al(H2O)3(OH)3] + -OH → [Al(H2O)2(OH)4]- + H2O
Aluminium compounds of weak acids are extensively hydrolysed to [Al(H2O)3(OH)3] and the corresponding hydride, e.g. AlN → NH3.
The Other Metals:
Gallium is also amphoteric, and its oxide Ga2O3 is slightly more acidic than Al2O3. However Indium is more basic than Ga and only weakly amphoteric, and will not dissolve in aqueous alkali. This is again related to the electronic and size factors mentioned.
Thallium behaves as a moderately strong base but is not really comparable as it normally exists as Tl(I) not Tl(III) (Tl(III) is strongly oxidising). Tl metal tarnishes in air and reacts with water to give TlOH.
Unsurprisingly, Tl(I) compounds show many similarities to the alkali metals’ compounds – TlOH is very soluble and a strong base, Tl2CO3 is very similar to M2CO3 where M = Na, K.
Range of Oxidation States –
The standard electrode potentials of the heavier Group 13 elements reflect the decreasing stability of the +3 oxidation state in aqueous solution, and a tendency to form +1 compounds instead (particularly Tl).
The stability of the +1 state in Group 13 increases in the sequence Al < Ga < In < Tl. The occurrence of an oxidation state which is 2 less than the group valency is referred to as the “inert pair effect” and is a common effect amongst the post-transition metals.
The explanation is that there is a decrease in bond energy with increase in size from Al to Tl so that the energy required to involve the s electrons in bonding is not compensated by the energy released in forming the 2 additional bonds. This is hard to quantify, but the net result is that the higher oxidation state becomes progressively less stable with respect to the lower oxidation state as atomic number increases within a group.
It is noted that an element is more electropositive in its lower oxidation state than in its higher, i.e. the lower oxide and hydroxide are more basic, while the higher oxide and hydroxide are more acidic. This is due to factors such as ionisation energies, cation size, hydration enthalpy / entropy. Thus for [M(H2O)x]n+, the larger n, the more likely it is to donate a proton.
As a consequence, the electronegativity trend of decrease down the Group is now reversed, and increases from Al to Tl.
Types of Compounds –
Hydrides
Distinctly less cluster compounds when compared to Boranes. AlH3 is a colourless non-volatile solid which polymerises via Al-H-Al bonds. It is a strong reducing agent and reacts violently with water and protic solvents to liberate H2. There are, as with boranes, 3c2e bonds present (no metal-metal bonds present at all). It can be prepared by combining LiAlH4 and AlCl3 in ether.
LiAlH4 is far more stable than AlH3 and can be used as a reducing agent with great versatility in an ether solvent. It is prepared by combining LiH and AlCl3.
Another well-known hydride is Al(BH4)3, the first fluxional compound found.
Gallane, GaH3, is even harder to prepare than the aluminium analogue. It decomposes spontaneously at room temperature. In the vapour phase its structure is similar to diborane (Ga2H6) but this aggregates in the solid phase to (GaH3)n. It reacts in a similar way to diborane, e.g. amine cleavage. Interestingly, ammonia cleaves asymmetrically to give [H2Ga(NH3)2]+[GaH4]-, whereas NMe3 cleaves to give (Me3N)2GaH3.
As the trend would indicate, InH3 and TlH3 are too unstable to exist in the uncoordinated state, and even LiMH4 compounds decompose at 273 K (where M = In, Tl).
Halides
These metals are capable of both +1 and +3 oxidation states, therefore we would expect to see monohalides as well as trihalides (unlike boron where there are no monohalides, but there are cluster compound possibilities).
All the monohalides are known, but AlH is a short-lived diatomic species. This would appear to be unusual given that the Al-H bond is the strongest, but the compound is not stable simply because it favours the +3 state too greatly and disproportionates.
Aluminium
The Aluminium trihalides can be made by direct combination of the elements (with the exception of fluorine where HF is used).
AlF3 in fact has unusual properties when compared to the other AlX3 compounds. It is involatile and insoluble, and has a much greater heat of formation. This is attributed to a change of coordination number (with X = F Al coordinates 6 atoms, while with X = Cl it drops to 4). This is a common trends amongst the post-transition metals. AlF3 has a structure related to Rhenium Trioxide and it is a fairly open structure (possibility of hydrates).
AlCl3 is actually 6-coordinate as a solid, but on approaching the liquid phase decreases to 4 – a molecular dimer of Al2Cl6. There is a corresponding increase in volume on melting, and a drop of conductivity to almost zero (clearly a change to covalent bonding). In the gas phase the dimers persist, but at high temperature decomposes to AlCl3.
Al2Br6 and Al2I6 exist in the crystalline phase as well as liquid, and they form AlX3 in the gas phase.
The Al trihalides form a large number of addition compounds or complexes such as in the Friedel-Crafts reactions where they are catalysts. Hydrocarbons bind weakly to AlX3 while Nitrogen binds strongly. Alkyl halides are studied extensively due to the aforementioned reaction, and it is found that the halide binding strength varies with how strong the R-X bond is. This can be probed using vibrational spectroscopy.
Other Metals
As with AlX3, the trifluorides of Ga, In and Tl are non-volatile and have high melting points and heats of formation. They show similar divergence from expected properties when compared to Boron and Aluminium – the factors of electrode potential, ionisation and electronegativity determine this. For example, the trihalides become less stable down the group, in direct opposition to the expected trend based on Group 3 trihalides.
These trihalides show a greater tendency to form addition compounds MX3Ln (n = 1,2,3). For example, the MX4- ions persist in ethereal solution, though some have a tendency to coordinate water additionally in aqueous solution.
Thermochemically, Ga and In differ from B and Al in having an underlying d10 configuration which can in principle take part in dπ-dπ back bonding with donors such as S (not O or N). Trends can also be explained by differing polarisabilities, where Ga and In are described as class-b (“soft”) acceptors whereas B and Al are class-a (“hard”). This is however a description, and the explanation lies in the MO’s of the donor and acceptor orbitals.
It should be noted that trihalides of Thallium are considerably less stable than the others in Group 13, and TlF3 will rapidly hydrolyse to Tl(OH)3 and HF. It does not form TlF4- in solution, and MTlF4 compounds have structures quite different (similar to fluorite structure – 8 coordinate). TlX3 where X = Cl, Br can be obtained indirectly by first making the tetrahydrate and dehydrating with SOCl2. TlI3 is highly unusual, and contains the linear I3- ion (like CsI3) and is clearly Tl(I) as opposed to Tl(III).
The Lower Halides of these metals are more stable than AlX. GaF and InF are however only stable in the gas phase, but as a rule stability of the monohalides increases with increasing size of the anion and GaI is quite stable, melting at 271oC. Again there is the possibility for addition compounds which can actually give rise to apparent “dihalides” such as GaI[GaIIIX4].
As expected, Tl monohalides are actually more stable than its trihalides due to the preference for the +1 oxidation state. TlX where X = Cl, Br, I are excellent insulators with negligible conductivity and an energy gap between the valence band and conduction band of about 3eV. Forcing the lattice to close together either by pressure or heat reducing this energy gap leads to metallic conduction as the two bands start to overlap.
Oxides and Hydroxides
Al2O3 (corundum) is an oxide ore of aluminium with some very useful properties – it is extremely hard, non-volatile and has high melting point, as well as being chemically inert and a good insulator. It is used as an abrasive and in ceramics as well as in production of Aluminium metal. Aluminium oxides are also present in most of the precious gemstones. The α-M2O3 structure comprises a HCP array of oxide with two-thirds octahedral holes filled with M. This is adopted by Ga2O3 as well. There is also a less compact cubic form, γ-Al2O3.
Aluminium Hydroxide itself does not occur in nature, but can be made by precipitation from alkaline solutions to form Bayerite, α-Al(OH)3. The γ-Al(OH)3 (Gibbsite) is more stable though, and is formed on heating the alkaline solution.
Gallium oxides are structurally very similar to those of Aluminium, while Indium oxides and hydroxides tend not to form the layer structures. Thallium is, as expected, quite different due to the preference for +1 Oxidation State, and Tl2O is known. Tl2O3 does exist, however, but it is not the predominant form and tends to be hydrated.
Aluminium also forms a range of mixed phase oxides, such as spinel (MgAl2O4). These are of the form AB2X4.
Chalcogenides
These are not as stable as the oxides for Aluminium, although Al2Y3 where Y = S, Se & Te are all known. They hydrolyse rapidly to give Al(OH)3 and H2Y. The small size of Al leads to tetrahedral coordination and their structure is usually related to wurzite (ZnS).
Unsurprisingly the chalcogenides of Ga, In and Tl are much more numerous, and often studied in relation to semiconductors.
Interestingly, GaS exists, but as opposed to +2 oxidation state, has Ga-Ga bonds present so that each Ga is coordinated by 3 S and 1 Ga. The same is true for GaSe and GaTe. InS and InSe are also the same, but InTe and the TlY compounds adopt a structure of MI[MIIIY2].
Compounds with Group 15
These are known, but less varied. They tend to adopt the cubic ZnS structure unless highly ionic. Thallium does not form simple compounds as they tend to explode. There has been recent study into Group 13 / Group 15 compounds as they have applications as III-V semiconductors (when the constituent elements are reacted at high temperature and pressure to form MIIIXV compounds).
For these III-V compounds, as X gets larger (down Group 15) there is a tendency for a lower melting point and energy gap.
Moving down Group 13, MN becomes more reactive, with AlN being inert to acid and alkali, while GaN decomposing in alkali and InN decomposing in both. MP and lower Group 15 compounds decompose more easily.
Organometallic Compounds
Aluminium has a very wide range of organometallic compounds, many of which are used as stereoselective or synthetically controllable reducing agents. Also worthy of note, the trialkyls dimerise just as AlH3 and AlCl3 do to give Al2Me6 molecules.
Ga, In, and Tl do not form as thermally stable compounds, nor do the trialkyls dimerise. The M-C bond also becomes less reactive down the Group (although Ga and In are approx. equal). Thallium in particular gives very stable organometallics such as R2TlX (air stable, non-hydrolysing), and TlMe2+ is stable in solution.
GaR3 can be made either by adding HgR2 to Ga, or by adding RMgBr to GaCl3. GaR3 is low-melting, mobile, and flammable. In and Tl form similar compounds but tend to have higher melting/boiling points.
GROUP 14
Carbon –
Occurrence –
Elemental carbon, organic compounds, CO2, carbonates.
Elemental Form & Allotropes –
Diamond – 3D structure related to Zinc Blende. An insulator. Tetrahedrally coordinated carbon atoms as a cubic cell (FCC).
Graphite – 2D layered structure made up of hexagons. Conducts. There are two forms depending on ABABAB (α) or ABCABC (β).
α-Graphite is more thermodynamically stable than diamond. Diamond can however be made from graphite by applying a very high pressure (it needs to be high enough to move atoms).
Buckminsterfullerene – C60 molecules.
Graphite Intercalation Compounds:
Atoms or molecules fit in between the layers (one layer at a time). The stoichiometry will be KC8, KC20 etc. Hal2 also does this. This process increases conductivity (K donates electron to band, Hal accepts electron and produces e-holes).
In fullerenes, K3C60 is a superconductor at 18K. They have alkene-like character in reactions (not benzene).
Reactivity –
Carbon has much higher ionisation energies than the other Group 14 elements.
Diamond is obviously very unreactive at room temperature. Graphite is a little more reactive due to its layer structure allowing some locus of attack (for example, it can be oxidised by hot conc. HNO3).
At higher temperatures, Carbon will react with H, F, O, S, Si and B, plus many metals. It is an active reducing agent, often used in industrial processes to reduce metal oxides.
The fullerenes undergo a few unusual reactions. They can be hydrogenated, and undergo oxidation with halides and O atoms, as well as be reduced to fullerides. Addition in a similar way as to olefins can also occur.
Range of Oxidation States –
[He]2s22p2 – oxidation state of +4, bonding is maximised. Ionisation energy is very high and C4+ very small, so C4+(X-)4 very unstable with respect to CX4. Thus, no cationic chemistry, except:
Types of Compounds –
As well as the obvious organometallic compounds, which comprises an entirely separate field, there are a range of possible compounds that carbon is part of.
Carbides
Carbon forms binary compounds with most elements. Carbides can be prepared by:
- Direct combination of the elements at high temperature,
- Reaction of metal oxide with Carbon at high temperature,
- Reaction of heated metal with gaseous hydrocarbon,
- Reaction of acetylene with electropositive metals in liquid ammonia.
Carbides are typically either highly ionic (M is early Group 1 & 2 and early TM’s), highly covalent (non-metals), or interstitial (middle TM’s). Some metals are unable to form stable carbides, namely Groups 11 & 12, the platinum metals, the post-transition metals of Group 13.
Carbides will tend to undergo hydrolysis, e.g. CaC2 + 2H2O → Ca(OH)2 + C2H2, which is highly exothermic.
Carbon Hydrides
More commonly known as hydrocarbons – very vast topic! Illustrates the ability of carbon to catenate into very long chains.
Unsaturated hydrocarbons such as ethene are effective ligands.
Methane is the only hydrocarbon thermodynamically stable wrt its elements. This can be compared with for example the boranes, where heating them leads to more complex boranes being generated would instead lead to the simplest hydrocarbon of all being formed – CH4.
Carbon Halides
CF4 is extremely stable, with a melting point close to CH4. It is thermally stable and chemically inert due to the great strength of the C-F bond, and hence fluorocarbons resist attack from acids, alkalis, oxidising and reducing agents extremely well, even up to 600oC.
CCl4 is often used as a solvent, but it is toxic. It is also used as the starting material to make other chlorofluorocarbons, formerly used in aerosols and as refrigerants.
CBr4 however is much less stable than the lighter tetrahalides, and is a solid at room temperature. The trend continues, and CI4 is even less stable, and also a solid.
Carbon Oxides
CO and CO2 are both very stable oxides, but there also exists 3 other oxides of lower stability – C3O2, C5O2 and C12O9.
C3O2 is a yellow solid, made by dehydrating malonic acid. It is a linear molecule, essentially: O=C=C=C=O. Thermolysis gives C5O2, which has similar properties.
Both CO and CO2 have very strong C-O bonds which confers thermal stability, but they are both quite chemically reactive. They are both very weak acids.
CO reacts with hydroxide at high temperature to give acetates.
Carbon monoxide has weak dipole with the negative on carbon due to π donation. It is actually a weak base. Bonding to a metal is possible due to π back donation again, but the metal needs to be a strong electron donor (low oxidation state). When this occurs, the C-O bond is weakened and easier to stretch. CO as a ligand allows it to react more easily, for example in reducing it to give methanol, or carboxylation of methanol to give acetic acid.
Synergic Bonding
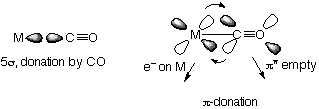
CO can also be used as a reducing agent in the extraction of metals (blast furnace).
CO2 is less volatile than CO, and it uses are usually for its physical properties as opposed to chemical (e.g. refrigerant, inert atmosphere, carbonating agent for drinks).
Its main studies have been in using and generating 14C for carbon dating and tracers, and in interpretation of the acid-base equilibrium on dissolving it in water:
H2CO3 ⇌ CO2 + H2O which depends greatly on pH.
CO2 can also act as a ligand, but it is less common. The carbonate ion, CO32-, on the other hand, is common and often a bridging donor.
Chalcogenides
CS2 is stable analogue of CO2, but CS is an unstable reactive radical even at low temperatures. More complicated chalcogenides can be made, usually by reduction of CS2.
CS2 itself is made by combination of sulphur and methane at high temperature. It is a poisonous gas, and used in manufacturing other chemicals (such as halogenation to make carbon halides).
CS2 is more reactive than CO2, and more readily forms complexes as a ligand, and also performs insertion reactions.
Carbon-Nitrogen
CN- is a very strong ligand, and isoelectronic with C22-, CO, N2 and NO+.
HCN is extremely poisonous and has a very high dielectric constant. It dissolves in most solvents. It is a very weak acid (weaker than HF) and produced by catalytic reaction of CH4 and NH3.
Metal Cyanides can be produced from it by neutralising HCN, and their crystals usually have NaCl or CsCl structure.
Cyanogen, (CN)2, is poisonous like HCN, and when pure is very thermally stable (impurities tend to cause it to polymerise).
CN- as a ligand is ambidentate, either donating from C or N depending on the metal’s acceptor orbitals. It is for this reason also that it is frequently a bridging ligand.
Silicon
Occurrence –
Very abundant (2nd only to Oxygen), and has uses in building materials, glasses, ceramics.
Typically found as SiO2, as 4-coordinate units {SiO4}. Never Si(s). Si can be obtained by reducing quartzite or sand over coke.
Extremely pure Si (for semiconductors) is obtained from SiCl4.
Si usually adopts the diamond structure, and is so regular when pure that it has been used to get the most exact measurement of Avogadro’s Number to date.
Elemental Form & Allotropes –
Long chains (catenation) are unstable with respect to SiH4 due to weak Si-Si bonds (unlike carbon). This is a trend down the whole group.
Reactivity –
Silicon is more volatile than C and has a much lower energy of vaporisation (Si-Si is weaker than C-C). Elemental Si is unreactive except at high temperatures, even to Oxygen, for which it has a very high affinity. This is probably because a fine film a few atoms thick of SiO2 forms on the surface, which is unreactive to the remainder. Temperatures of over 900oC are required to form SiO2 from O2 and 1400oC to react with N2.
Si will resists all but the strongest acids, but reacts with alkali, forming SiO42-. It also reacts with halogens (presumably the SiO2 barrier does not prevent this).
Si will form binary compounds with C, but not with the other Group 14 elements. Si is usually 4 coordinate in all compounds, although there are occasional exceptions.
Multiple bonds are also not favoured, since Si is larger. No O=Si=O. Polymerises instead – internal bonds instead of intramolecular. Large core means poor overlap, so weak π-bonds. Si=Si compounds are known, but need to be stabilised by sterically very bulky ligands.
SiX4 compared to CX4 (X= Halogen).
SiX bonds are polar, Si is large and has vacant 3d orbitals, so reactive. Will explode with O2.
SiF4 + HO- → SiO2, SiF62- etc… (unlike CF4)
SiO → Si + SiO2
Types of Compounds –
Silicides
As with borides and carbides the formulae of metal silicides cannot be rationalised by the application of simple valency rules, and the bonding varies from metallic to ionic and covalent. There are a large range of possible stoichiometries, as was seen for the borides.
Silicon is more electropositive than carbon (like boron) and structurally the silicides are more similar to borides than carbides. However, the size of silicon compared to boron prevents any isostructural compounds. There are however very many possible silicides (most of Groups 1-10 will form them) with the greatest range of stoichiometries shown by the Transition Metals and Uranium.
The silicides of Groups 1 & 2 are actually more reactive than those of transition metals (unlike carbides and borides), and they hydrolyse to release hydrogen gas, or form silanes in acid.
Hydrides
These are more typically called silanes, and are of the form SinH2n+2 as for simple hydrocarbons. They can form chains up to n = 8 and rings up to n = 6, but Silicon’s ability to catenate is no where near as stable as carbon due to its size as mentioned. In fact SiH4 is the only silane stable indefinitely at room temperature, as Si2H6 decomposes (albeit very slowly) and tetrasilanes and above decompose quite rapidly.
The silanes are less volatile than the boranes and hydrocarbons, but more so than the germanes. They are also considerably more reactive than hydrocarbons, and this is due to the larger radius of Si facilitating attack by nucleophiles, more polarised Si-X bonds, and the presence of low-lying d orbitals which permit 1:1 and 1:2 adduct formation (lower activation energy). The relative bond energies are also important, with Si-Si < Si-C < C-C and Si-H < C-H. When X are other elements C-X < Si-X though.
Halides
The compounds of SiX4 are volatile, colourless and reactive. They also show very low melting / boiling points when compared to Aluminium halides in particular (AlF3 = 1291oC while SiF4 = -90oC). This is not however due to a change from ionic to covalency as might be expected, but instead due to a structural change in the lattice, since [AlF6] units infinitely repeat while [SiX4] is discrete amongst the lattice.
Interestingly, a greater degree of catenation is achieved for silicon halides compared to silanes, which is the opposite of carbon’s properties. This is due to additional back-bonding from the filled halide pπ orbital into the empty Si dπ.
Silicates
Silica, SiO2, is a widely studied compound and has many, many polymorphs even when pure. It is mostly found as α-quartz.
The structure of the silicon oxides is most stable when [SiO4] units are corner-shared.
SiO2 is essentially unreactive, but it will react with alkali (but not acid).
Silicates can develop chains or be discrete, depending on the sharing of the O atoms.
Other Compounds
SiS2 can be made from direct combination of the elements under heat, but it is quite reactive and hydrolyses and reacts with ammonia readily. Its structure is also not nearly as varied as those of SiO2.
Si=S bonds can also be formed (as can Si=N), but they require high temperatures and are not very common.
Si + N2 gives a useful crystal, Si3N4. This is one of the most chemically inert compounds known, and it retains its strength, shape and inertness even at 1000oC, as well as being a good insulator.
Further evidence of dπ orbital contributions are found when looking at other nitrogen compounds, such as N(SiH3)3, which is planar (analogous to BX3), as this is the only way to maximise the pπ-dπ back-bonding.
It should also be noted that there are a wide range of organosilicon compounds found. This is attributed to the strong Si-C bond (stronger than Si-Si in fact, and almost as strong as C-C).
Other Group 14 Elements – Germanium, Tin and Lead
Occurrence –
Germanium is typically found as a trace element in other ores, particularly alongside Zinc, with which it shares amphoteric properties. For this reason it can be hard to separate the two, especially since Germanium is found in such small quantities.
Tin is typically found as SnO2, which can be reduced over coals (which has been known for some time!). The main problem with extraction is iron impurities.
Lead is the most abundant heavy metal, as it is the end product of many radioactivity series’. It is found mostly as PbS, but also as PbSO4 and PbCO3. It is extracted from PbS by froth flotation then roasting to give PbO, then reduction over carbon.
Elemental Form & Allotropes –
Germanium is a brittle, grey-white solid with diamond structure. It is a metalloid like Si and has a similar resistivity.
Tin has two main allotropes at room temperature. The most stable is a white, tetragonal β-Sn, but at lower temperatures this tends to a grey α-Sn which has the cubic diamond structure. Interestingly, the density increases for the β-Sn (at higher temperature). This is due to and increase in coordination from 4 to 6 (even though Sn-Sn lengthens).
Lead is a blue-grey soft metal with high density (twice that of Sn and Ge and 5 times that of Si).
Reactivity –
Trends in Group 14 are similar to those in Group 13, in that there is pairing of ionisation energies with Si and Ge being similar, followed by Sn and Pb being similar. This is for the same reasons as in Group 13 – related to the filling of the 3d and 4f shells causing contraction of radius. There is also a trend of decrease melting / boiling points down the Group.
Ge is more reactive and more electropositive than Si. It dissolves in strong acids, but is unaffected by water or dilute acid/alkalis unless a strong oxidising agent is present. Fused alkalis will react to give germanates. Ge will react with O2, S2 and halogens with gentle to moderate heating.
Following the trend, Sn is even more reactive and electropositive than Ge, though it still exhibits amphoteric properties. It is stable in water and air, but reacts with steam and oxygen upon heating. Dilute acids most be strong acids in order to react (e.g. HNO3). There is a greater tendency to form SnII compounds in this case, e.g. with hot H2SO4 SnSO4 forms. Hot alkalis give Sn(IV) compounds. Sn also reacts easily with halogens X2.
Unsurprisingly Pb is reactive (in fact it is pyrophoric), but this is diminished due to a fine oxide coating that forms (in a similar way to SiO2). As a result high temperatures are needed to form PbO in air. It does tend to react with acids however, especially HCl, HNO3 and acetic acid (a problem for containers for wine and fruit juices).
Catenation can again occur for these compounds, although it is far less pronounced than for C and Si. This is due to a decrease in M-M bond strength. M-X bonds do decrease in strength down the group, but on the whole not by a huge amount (apart from M-H).
It should also be noted that Sn and Pb have a tendency to form low oxidation state cluster compounds, and this trend for heavy metals continues into Groups 15 and 16.
Range of Oxidation States –
The inert pair effect, which has a consequence of stabilising the +2 oxidation state over the +4 down the Group, again applies here. However, there is a notable exception in the organometallic chemistry of Sn and Pb, which is almost entirely in the +4 state.
Types of Compounds –
Hydrides
Germanes (GenH2n+2) are colourless gases or volatile liquids for n = 1-5, and their physical and chemical properties are very similar to silanes, although they are all less volatile, and (unusually) less reactive.
GeH4 behaves as an acid in liquid NH3, and also reacts with alkali metals in this solvent. The hydrohalides are however considerably more reactive, and are made by adding HX to the appropriate starting compound.
Tin Hydrides are far less stable, and even SnH4 decomposes slowly at room temperature to its constituent elements. Sn2H6 is known but not very stable, and both act as strong reducing agents.
PbH4 is almost unknown, even at low temperatures. It is however possible to form alkyl derivatives.
Halides
Ge, Sn and Pb will form halides in both the +2 and +4 states. Pb(II) halides are more stable than Pb(IV), while the opposite is true for Ge and Sn, consistent with the steady increase in stability of the dihalides and the +2 state in general down the Group. Complex halides are also known for both oxidation states.
For Ge, the tetrahalides hydrolyse readily and are important intermediates in organogermanium compounds. The dihalides hydrolyse to give Ge(OH)2. Complexes such as GeX62- are also known where X = F, Cl.
For Sn, there are a wider range of stable dihalides compared to smaller Group 14 elements. The lone pair present on the Sn(II) also causes some unusual stereochemistry to be observed. They are typically mild reducing agents. The tetrahalides show less unusual stereochemistry, although some of their properties differ from the other Group 14 elements encountered. For example, SnF4 is highly hygroscopic, and forms extended polymeric structures unlike GeF4, SiF4 and CF4.
For Pb, the dihalide is much more stable the tetrahalide as expected. In fact, the only stable tetrahalide is PbF4 – even PbCl4 decomposes unless kept well below room temperature. It should be noted that there is less stereochemical variety in the dihalides of Lead – this is because of the greater size of the lead ion (Sn and Pb compare very closely to Mg and Sr).
Oxides
For Germanium, GeO2 is much more stable than GeO, which disproportionates, although GeO can be prepared by reducing GeO2 or dehydrating Ge(OH)2. There is quite a bit of similarity between Ge(IV) and Si(IV) oxide chemistry.
SnO, on the other hand, exists in several modifications. It is usually prepared by alkaline hydrolysis of Sn(II) salts, and then dehydration in the absence of air. SnO2 can be formed by heating in air, but heating in the absence of O2 causes disproportionation as for GeO. There is also the possibility of mixed oxides, although the only well-characterised one is SnII2SnIVO4.
SnO is amphoteric, and dissolves in both acids and alkalis readily to give either SnII complexes in acid, or Sn(OH)3- in base. Intermediate pHs lead to condensed basic oxide-hydroxide species, e.g. [(OH)2SnOSn(OH)2]2-.
SnO2 is, however, the main ore of tin and it has the rutile structure. It is insoluble in water and dilute acid/alkali but dissolves readily in fused alkali hydroxides to form stannates – MI2Sn(OH)6.
Sn(OH)4 is not known, but hydrated SnO2.H2O can be formed.
PbO has a number of structural forms, though the tetragonal form is the most stable and widely used. There is also a mixed valency Pb3O4 compound made by heating PbO. PbO2 does exist and has the rutile structure, but decomposes on heating, eventually to PbO although it proceeds via a number of cluster-like lattices.
In terms of hydroxides, there is no simple hydroxide of Pb. This is likely due to the increasing pH of solutions of PbII salts leading to hydrolysis and condensation to give something like [Pb6O(OH)6]4+.
Other Compounds
Oxoacids are also worthy of note, although for Germanium they are largely unstable. For Tin however there are numerous oxoacid salts of SnII and SnIV. Pb also forms stable oxoacids, mostly as PbII, although PbIV(OAc)4 is a well-known oxidising agent in organic chemistry.
All the MX chalcogenides are known (X = S, Se, Te). GeS and SnS have a layered structure similar to the isoelectronic black Phosphorus. GeS is prepared from GeS2, while PbS and SnS are made from MII salts directly.
It should also be noted that although Ge, Sn and Pb do not undergo extensive catenation, they will form cluster compounds readily.
In terms of organometallics, organogermanium compounds are not nearly as thermally stable as organosilicon compounds, and often far more reactive. Organotin reagents tend to find more uses in organic chemistry, especially in radical mechanisms, while Lead Acetate is often used as an oxidising agent as mentioned. Lead organometallics however are less extensive than for Tin, and unusually are almost always Pb(IV) as opposed to the usually more stable Pb(II).
GROUP 15
Nitrogen –
Occurrence –
N2 gas comprises most of the atmosphere. There is not much nitrogen in rocks (mostly nitrates). It is usually obtained by distilling from air.
Elemental Form & Allotropes –
Forms molecular N2 gas – hard to dissociate. Solid not obtainable – close to absolute zero.
Reactivity –
Very strong NN bond, but N-E bonds commonly weak. Compare weak N-N single bond, due to non-bonded electron repulsion. Thus, many nitrogen compounds are thermally unstable with respect to N2.
N2 is inert, has no bond polarity, and has a large HOMO→LUMO gap, so thermally very, very stable. Many nitrogen compounds are explosive wrt N2 as final product, e.g. azo compounds.
At high temperature, N will combine with Groups 1 & 2, + B, Al, Si, Ge to form nitrides.
N3- is a good ligand (nitrido), particularly to 2nd / 3rd row Transition Metals. It is a very strong π-donor.
Catenation is usually limited to 3xN, but occasionally –N-N=N-N-N- can be formed.
Range of Oxidation States –
[He]2s22p3 and oxidation states -3→ +5. More valence electrons than orbitals plus lone pairs. Aqueous redox chemistry, and high oxidation states are oxidising.
Types of Compounds –
Nitrides
N forms binary compounds with almost all elements (not noble gases). The nitrides divide roughly into four groups –
- Salt-like – Li3N and M3N2 (M = Group 2). Group 1 does not form nitrides due to sterics (form azides).
- Covalent – (CN)2, P3N5, S4N4.
- Diamond-like
- Metallic (interstitial) – metal lattice with holes filled by N (most T. Metals). These are hard, opaque and inert, and conduct electricity. They are in many ways similar to the carbides and borides.
Group 13 MN compounds do not really fit into this classification.
Hydrides
NH3 is the only thermally stable hydride. Prepared industrially by the Haber process (500oC, high pressure), and naturally by nitrogen fixation.
There is extensive H-bonding, though it has low density, low viscosity and low conductivity. It has a high dielectric constant, though significantly lower than water.
Ammonia will burn, but reluctantly. If done over a Pt (or similar) metal, nitrogen oxides are formed.
Its typical use is as a non-aqueous ionising solvent (allows use of strong redox agents). Allows preparation of compounds in low Oxidation States, or those that are unstable in water.
Other hydrides possible are N2H4 and N3H, as well as anions and cations of both. Ammonium Hydride, NH5, is not known.
Also, NH3 + NH2Cl N2H4 + NaCl + H2O (Raschig process)
But, 2NH2Cl + N2H4 N2 + NH4Cl
The structure is most stable as:

NH3 basic. N2H4, weakly basic
NH4+, N2H5+, N2H62+
Both NH3 and N2H4 mild reducing agent (ox states -3 and -2).
Ammonia salts have good solubility.
N2H4 is physically similar to water and ammonia due to H-bonding, and has a large dipole (no trans form). It is stable kinetically but an endothermic compound. It burns rapidly and completely, and a stronger reducing agent.
NH2OH is thermally unstable and hygroscopic. It is usually handled as a salt or in solution (where it protonates to a more stable form).
HN3 is explosive in anhydrous form, and unstable. It is actually used in detonators. It is also a deadly poison. It is not linear symmetric N-N-N as in M+(N3)-.
Halides
Maximum coordination is 4, but charged NF4+.
NI3 is not known, and NCl3 and NBr3 are thermally unstable and explosive.
NF3 is stable. It is unreactive, like CF4. Also a very poor ligand. It has a lower dipole moment than NH3 since the N→F dipole is reverse of N←H + lone pair. It is a weaker base.
NH3 + F2 → NF3 + N2F4 + N2F2
F-N3 is very unstable and highly explosive (it is essentially a covalent azide).
N2F2 is the analogue of hydrazine, and similarly is more reactive than NF3. It also dissociates to NF2 radicals in the same way as N2O4.
NH2Cl + HOCl → HNCl2 +H2O + NCl3
NCl3 is thermally unstable, yellow oil, and explosive. It is highly reactive and easily hydrolysed.
Oxides
All oxides of nitrogen thermodynamically unstable with respect to N2 and O2. There are however 8 molecular oxides known.
N2 + O2 → no reaction (even at 3000oC).
N2O:
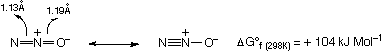
Nitrous Oxide is relatively inert even though unstable. This is due to an unusual decomposition pathway via a forbidden singlet-triplet transition (spin is not conserved).
NO:
NaNO2 + H2SO4 NO
Cu + HNO3 NO
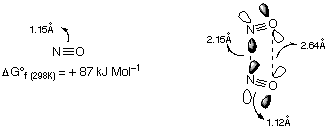
π*/π* overlap, giving long and weak N-N bond, diamagnetic.
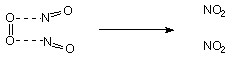
NO is a colourless gas, and is unusual in that it has an odd electron, but is stable. It is of course paramagnetic. The bond order is 2.5, with 1 electron delocalised around the whole molecule. Thus, it does not dimerise.
Like CO, can act a π-acid, and is a good ligand (nitrosyl) with many complexes known. There is a wide range of geometries possible (bridge, linear, bent for example).
N2O3:
NO + N2O4 N2O3
N2O3 readily dissociates above -100oC:
N2O3 NO + NO2
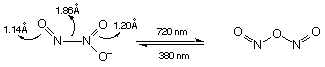
The symmetric and asymmetric isomer can be readily interconverted. It is blue.
Like (NO)2 dimer, long and weak N-N bond
N2O4:
Pb(NO3)2 PbO + 2NO2 + 1/2O2
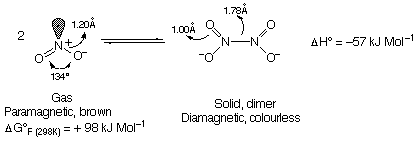
NO2 has unpaired electrons like NO, but these are less delocalised (mostly on N) and hence dimerises rapidly.
N2O4 is planar.
N2O5:
HNO3 + P2O5 N2O5
Structure
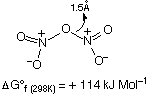
NO2+NO3- in solid or acids such as H2SO4
Oxoacids are also known, but most are unstable in free state. HNO3 is the main acid (HONO2), but also HNO2 is quite stable, although less strong acid.
Phosphorus -
Occurrence –
As phosphates (11th in abundance). These are common in living things. P can be extracted from phosphates by heating with carbon and silica.
Elemental Form & Allotropes –
Phosphorus has a very wide range of allotropic forms. They all melt to give tetrahedral P4 units, and this vaporises initially to P4 (g), but when very hot P2 (g) will form (this compared to N2 (g) all the way to well below room temperature).
α-P4 is known as white phosphorus. It is the most common form, and has a cubic array. It is actually the most reactive and volatile, even though common. Oxidation causes slow phosphorescence.
β-P4 is also white, but this time hexagonal. Both P4 allotropes are insoluble in aqueous solution, but highly soluble in CS2, NH3 etc.
Black Phosphorus is thermodynamically the most stable form. It is highly polymeric and dense. It is also a semiconductor, but is usually too impure to be used.
Red Phosphorus is often the most used in reactions. It is denser, has a higher melting point and is less reactive, and can be formed by heating white phosphorus in the absence of air.
Both red and black phosphorus show a much greater degree of catenation than white, and much more than nitrogen is capable of. They are as a consequence less reactive.
Reactivity –
This depends on the allotrope, as mentioned. Phosphorus is typically spontaneously chemiluminescent on reaction with moist air.
The N2 triple bond is far far stronger than that of P2, which explains the elemental forms. However, P-P is stronger than N-N, so phosphorus tends to catenate far more.
Phosphorus reacts to form binary compounds with most elements (not Sb, Bi or noble gases). White Phosphorus reacts with water on heating, but Red Phosphorus will not.
Range of Oxidation States –
+3 and +5 states are known. There is also the possibility of low-lying d-orbitals being used, as for Silicon.
Types of Compounds –
Phosphides
There is a large number of stoichiometries, similarly to borides.
nM + mP → MnPm (with heating). These phosphides can be classified in 3 main ways:
- Metal-rich Phosphides – hard, brittle. High thermal and electric conductivity. They are thermally stable and chemically inert.
- Monophosphides – their shape is influenced by size and electronic effects.
- Phosphorus-rich Phosphides – lower melting point and stability. They are semiconductors as opposed to metallic. They exhibit catenation of phosphorus atoms.
Phosphides of electropositive metals (Groups 1 & 2 and lanthanoids) show a degree of ionic bonding, but there is still extensive metallic and covalent interactions. They are easily hydrolysed by acid or water to PH3.
Hydrides
Phosphine, PH3, is the most stable. However, unlike nitrogen hydrides, there are a greater range due to catenation.
PnH2n+2 (n = 1 – 9).
n = 1,2,3 can be obtained pure, but stability does decrease rapidly.
Phosphorus can form cyclic and condensed PnHn, PnHn-2, etc compounds, but they are not usually as thermally stable.
Again, Phosphorus Hydride, PH5 (phosphorane), is not known.
PH3 is highly poisonous and very reactive (not like ammonia). It is however more stable than AsH3. It has the same structure as ammonia (indicating H-bonding present) but is less soluble in aqueous solution (so it occurs to a lesser degree). It is a strong reducing agent. In aqueous solution, PH3 is unlikely to protonate or deprotonate. It is also a good ligand to Lewis Acids.
Halides
PX3, P2X4 and PX5 known for 11 out of the 12 possible compounds (no PI5). This is compared to the very limited Nitrogen Halides.
PX3 compounds are volatile and reactive. They have the expected pyramidal structure.
PF3 is a haem poison, and this is because it is a similar ligand to CO. It slowly hydrolyses in water, but more rapidly in alkaline (to give H3PO3).
PCl3 undergoes many substitution reactions, and is also the precursor to most organophosphorus compounds. It can also be oxidised to PCl5 relatively easily.
Diphosphorus Tetrahalides:
P2F4 + H2O → F2POPF2.
P2Cl4 → PCl3 (spontaneously decomposes).
P2Br4 not well characterised.
Pentahalides:
PCl5 is a molecular gas with trigonal bipyramidal structure, but as a solid forms [PCl4]-[PCl6]+. This shows that it is very close to the ionic / covalent borderline. This is further emphasised when looking at PBr5, which forms PBr+PBr4- solid. PF5 is a thermally stable, reactive gas.
Oxohalides are also easily formed. This is achieved by oxidising P(III) to P(V) with formation of a P=O bond (very strong – thermodynamically driven). As a result OPX3 is common.
Other Compounds
There are 6 binary oxides and 9 binary sulphides.
PO (the NO analogue) is not well defined, but abundant in interstellar clouds.
Oxides can be obtained by oxidation of P4, and in controlled conditions under oxygen, will form P4O6. Heating this further gives red phosphorus and P4On.
P4O6 is made up of [P3O3] units, but it is more common amongst the other oxides to form from [PO4] units (the phosphate).
P4O10 is the most common of these, and is formed by burning phosphorus in air. In the hexagonal form this reacts violently with water (so is often used as a dehydrating agent), but the polymeric form reacts slowly.
The other oxides are less well characterised.
There is however a wider range of sulphides, of which P4S3 is the most stable. They have a more diverse stereochemistry and structure. P4S3 retains the P3 rings from Red Phosphorus, while P4S7 shows a greater degree of catenation.
There are also possibilities for stable oxosulphides.
Phosphorus also forms the greatest range of oxoacids (rivalled only by Silicon), and this is due to the great strength of the P=O bond.
They are always of the form: O=P(OH)(R1)(R2), where the R groups are either hydrogens or P-O-P linkages. H3PO4 is the strongest of these acids (phosphoric acid).
Phosphorus-Nitrogen compounds are also known, and are essentially derivatives of the oxoacids. Thus P-H translates to P-NR2 and P=O to P=NR.
There are also many organophosphorus compounds, e.g. PMe3 and PPh3 which are used as soft nucleophiles and strong σ-donor ligands. There are also P(V) compounds like P(Me)5.
Other Group 15 – Arsenic, Antimony and Bismuth –
Occurrence –
Post-transition Metals, so as expected they occur predominantly as chalcogenides (with S, Se, Te). There are also arsenides, antimonides and bismuthides, giving:
As2S3, As4S4, FeAs2, CoAs, NiAs.
Sb2S3, Pb, Co, Ag ores.
Bi2S3, Pb, Co, Ag ores.
Elemental Form & Allotropes –
Antimony is interesting in that it occurs as two major isotopes, 121Sb and 123Sb split roughly 50:50. Bismuth is also interesting in that its only isotope, 209Bi, is the heaviest stable isotope possible – all subsequent elements in the Periodic Table beyond Bi (atomic number = 83) are radioactive and hence unstable.
There are again several allotropes for these elements, but not as vast a range as for phosphorus.
Arsenic is more volatile than Sb and Bi, and has 3 main allotropic forms. Antimony has 5, and Bismuth has a range of polymorphs that are hard to define.
Reactivity –
Arsenic and Antimony are metalloids. Arsenic is stable in air but oxidised surface is present. It has a poisonous odour. Heating it generates the oxide As4O6. It is not easily attacked by water, alkali or non-oxidising acids, but dil HNO3 forms arsenious acid (H3AsO3) and conc HNO3 gives H3AsO4 (arsenic acid).
There is no As3- ion due to coulombic repulsion, so Na3As exists as an intermetallic (alloy-like) compound. All these elements prefer to form strong covalent in a similar way to Phosphorus, although none are as strong. Covalent linkage strength decreases in the order: P > As > Sb > Bi, i.e. as the elements get larger. For example, BiH3 is unstable.
Antimony reacts in a similar way to As, but is on the whole less reactive. It oxidises on heating, but conc. oxidising acids are needed to generate Sb2O5 (hydrated).
Bismuth shows highly electropositive behaviour, and Bi2O3 is very basic. This is in comparison to SbOx and AsOx being amphoteric and NOx and POx being acidic.
Range of Oxidation States –
ns2np3 valence electrons, but again the core electrons affect trends considerably due to low-lying unfilled orbitals (not present in N) and filled d/f orbitals after the 3rd and 4th Periods. +3 and +5 Oxidation States are the main ones, although +5 gets less stable down the group such that Bi(V) is extremely oxidising.
Arsenic is especially against forming As(V), and this is a common theme amongst the p-block for elements in Period 3. This is because of the filled 3d orbitals giving rise to d-block contraction which lowers the energy of the 4s electrons (so they are less easily promoted to attain +5 oxidation state).
Types of Compounds –
Intermetallic Compounds and Alloys
Arsenides, Antimonides, Bismuthides – show a range of composition and stoichiometry. There are also many nonstoichiometric compounds.
With compounds of Group 13, form III-V semiconductors.
Hydrides
They are all very, very poisonous, and unstable colourless gases. BiH3 particularly unstable.
There is no H-bonding observed, and they also show low proton affinity (not often form NH4+). They also become less thermally stable down the Group.
They oxidise to give the trioxide and water, and lower hydrides are not really known (unsurprising).
Halides
Of the trihalides, all 12 are known. The melting point and density trends indicate variation in structure, and it has been found that AsI3, SbF3 and BiX3 all form extended solid structures, while the remainder are volatile molecular species.
They react with alcohols to form M(OR)3.
Only the fluorides form stable pentahalides, with the exception of SbCl5. These are formed by reacting fluorine gas with the element or its oxide. SbCl5 is formed by SbCl3+Cl2. There are no bromides or iodides since M(V) is too oxidising (in a similar way to Thallium (III) ).
The pentahalides again illustrate the instability of the +5 stable for the elements in Period 3 (after the 3d shell has filled), since AsCl5 is unstable, but PCl5 and SbCl5 (above and below it) are stable.
Oxides
Trend to basicity: As < Sb < Bi.
Trend to acidity: M(III) < M(V).
As2O3 can be formed by heating As in oxygen, or hydrolysing the trihalides. On heating to vapour, it forms As4O6. Its solubility is highly dependent on pH.
Interestingly, Bi(OH)3 is basic as opposed to As(OH)3 (arsenious acid).
For M(V) compounds, As2O5 has low thermal stability and is easily hydrolysed. MAsO3 compounds are also found. Sb(V) Oxides are poorly characterised, and Bi(V) Oxides even less so.
Sb(III)/Sb(V) and As(III)/As(V) have similar values as oxidising couples, while Bi(III)/Bi(V) is strongly oxidising, and capable of oxidising water to oxygen.
Other Compounds
There is a greater degree of M-M bonding occurring with these elements, and this is illustrated in the structures of the sulphides, which are numerous. It is also shown by the tendency to form cluster compounds.
There are also many stable oxoacid salts (NO3-, SO42-, etc). Their stability increases in the order As << Sb < Bi, presumably because Arsenic is insufficiently basic.
Finally, there is a range of organometallic compounds known, but their stability
is lower than those of phosphorus as the M-C bond is weaker (in the order As > Sb > Bi). Common
uses
in Organic Chemistry are as ylids (-M=CR2).
GROUP 16
Oxygen –
Occurrence –
Most abundant element on earth’s crust. Occurs predominantly as O2 (g), but also O3 in the atmosphere, and then oxides, silicates, carbonates and water on the earth. It is obtained by separation from air.
Elemental Form & Allotropes –
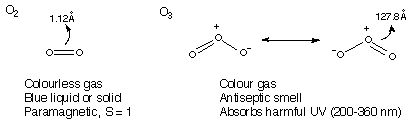
Two Allotropes: O2 and O3.
O2 is an odourless gas, blue liquid or solid (rotationally disordered), and paramagnetic (unusual – explained by MO, right).
O3 is a blue gas and black solid, diamagnetic and odoured. It is also bent, and more reactive. It is thermally unstable wrt O2.
Atomic O is found only in the ionosphere, and is very, very reactive.
Reactivity –
It is highly electronegative (only F greater), and highly reactive (oxidises). This is despite the high bond energy. Exothermic reactions can be spontaneous and explosive. High electronegativity, small size and p electrons make it in some ways similar to Nitrogen chemistry: H-bonds, covalency, pπ double bonds. However, there is more ionic variety as well.
Oxides form for all elements except He, Ne, Ar and Kr. Electron Configuration [He]2s22p4 → Oxidation State of O can vary from -2 to +2. Generally: +½ , 0, - 1/3 , -½ , -1, -2. It is usually -2 (-2 through to +2 are technically accessible). It can coordinate anything up to 4. E=O is common though, while 4 is uncommon.
|
Species |
Oxygenyl |
Oxygen |
Superoxide |
Peroxide |
|
|
O2+ |
O2 |
O2- |
O22- |
|
Bond Order |
2.5 |
2 |
1.5 |
1 |
|
Bond Length /A |
1.12 |
1.21 |
1.33 |
1.49 |
|
[p]*/e- |
1 |
2 |
3 |
4 |
|
Unpaired e- |
1 |
2 |
1 |
0 |
O2 is a π-acid and can behave as a ligand (as in haemoglobin). Complexing O2 can be useful, as it makes the oxygen molecule diamagnetic (no spin conservation required in reaction), and holds the O2 and reactant close together to lower activation energy. Coordinated O2 is often part reduced to O2- or O22-.
Lone pairs play important role in chemistry, as does aqueous redox chemistry as Oxygen is very oxidising (!).
Coordination Numbers (Stereochemistry)
- Unicoordinate O in E=O bonds very common.
- Two-Coordinate O ~ E-O-E groups are typically bent (VSEPR).
H3Si-O-SiH3 angle > 150o (π-interactions)
[Cl5Ru-O-RuCl5]4- linear M(dπ) ← O(pπ) bond
- Three-Coordinate O ~ typically pyramidal, e.g. oxonium ions, +OR3
~ trigonal planar in some OM3 groups (e.g. M = Cr).
pπ → dπ bonding.
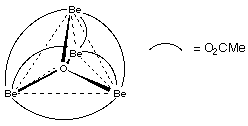
- Four-Coordinate O ~ Uncommon, e.g. basic beryllium carboxylates
Be4O(OAc)6.
Types of Compounds –
Hydrides
Water, H2O
Volatile, mobile liquid. Extensive H-bonding – high bp. Dipole. High Dielectric constant.
Self-ionization: 2H2O ⇌ H3O+ + OH- Kw = 10-14
Oxonium cation, H3O+ (isoelectronic with NH4+) - standard acid medium in water
Ubiquitous solvent - strongly solvates ions (due to dipole moment from Hd+-Od-).
Water as a solvent can also react – hydrolysis reactions. These typically generate hydrides from e.g. carbides, nitrides, etc. Also acid/base reactions are aqueous redox chemistry.
There are many phases of ice, depending on conditions. Hexagonal Ih is the normal form. Very low temperature gives Ic (cubic). Ih has lower density than the liquid due to regular H-bonding. Pure ice has low conductivity (impurities change this dramatically).
Crystallisation Interactions –
H2O coordinates in a cationic environment as e.g. [M(OH2)x], usually occurs where M is +2/+3 and highly coordinated.
H2O coordinates by H-bonding to oxoanions, e.g. CuSO4.5H2O, where 4 water molecules coordinate to the copper and the fifth H-bonds to the S.
Lattice Water – MF.nH2O. Especially large alkali metal fluorides form this, as water fills the lattice gaps due to the size difference.
Zeolite Water – framework silicates accommodate water molecules.
H2O2, Hydrogen Peroxide
Almost colourless liquid. More dense, viscous and less volatile than water. Forms H2O2.H2O in aq.
Hydrogen Peroxide, H2O2

Slight self-ionization; 2H2O2 ⇌ H3O2+ + HO2- K = 1.5 x 10-12
Lab Synthesis: BaO2 → BaSO4 + H2O2
The oxidation state of O is -1, so in between dioxygen and water. Thus it can be both an oxidising and reducing agent:
Oxidizing Agent: H2O2 + 2H+ +
2e- ⇌ 2H2O
Reducing Agent:
H2O2 is
itself reduced by stronger oxidizing agents, e.g. MnO4-
O2 + 2H+ + 2e- ⇌
H2O2
H2O2 is exothermic wrt elements, but decomposes: H2O2 → H2O + 1/2O2. This is normally slow, but accelerated by metals and alkali. It is explosive when anhydrous and pure – even a speck of dust can initiate.
Peroxides and peroxo-salts occur widely, e.g. peroxodisulphate (O3S-O--O-SO3)2-
Fluorides
OF2 is the most stable. It is a very poisonous, colourless gas. Pure form is stable up to 200oC. It is an oxidising and fluorinating agent.
HOF is hard to characterise as it decomposes to O2 + HF at rtp.
O2F2 is formed by direct combination at low pressure with electric discharge. It is similar to hydrogen peroxide in structure, but the O-O bond is shorter and O-F longer. It dissociates to radicals of F + OOF which dimerise to give F2On (n=1,2,3). It can be used as a high energy oxidiser.
Oxides
Readily combines at required temperatures. All elements except light noble gases.
They show a vast range of properties, e.g. from CO (hard to condense) to ZrO2 (mp = 3265oC). Also MgO = insulator, MiO = semiconductor, ReO3 = metallic. There is also the possibility for nonstoichiometric phases, typically with variable oxidation state transition metals. Thus there is no simple classification.
One useful classification is the Acid/Base one, although really this is the classification of the hydroxides.
ACIDIC: non-metal oxides, e.g. CO2, SO3.
BASIC: electropositive elements – Na2O, CaO, TlO, Ca2O3.
NEUTRAL: don’t interact with water, acid or alkali, e.g. CO, NO.
AMPHOTERIC: less electropositive, e.g. BeO, Al2O3, ZnO.
The amphoteric borderline moves further to the right of the Period as each Group is descended.
Acidity increases with oxidation state, e.g. MnO < Mn2O3 < MnO2 < Mn2O7.
Explanation:
Concept of "Polarizing Power" explains the trends.
Consider the reaction: EOn/2 +
(6 + n/2)H2O → E(H2O)6n+ + nOH-
~ En+ large (Group 1 and 2), E(H2O)6n+ ion stable, oxide is basic
~ En+ small, e.g. B3+, Si4+, => Hydrolysis → E(OH)n + nH2O neutral
e.g. Al(H2O)63+ is amphoteric Si(OH)4, or B(OH)3 are weakly acidic.
~ En+ very small, e.g. S6+, => further hydrolysis, and strong acid properties.
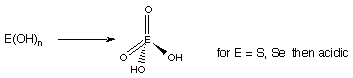
~ Explains why acidity increases with oxidation state.
Alternative viewpoint, as En+ gets smaller preference for:

Trends:
- Across a period, Zeff increases, bonding to O becomes more covalent and localised.
=> change from ionic → polymerized → small molecule structures
=> decrease in coordination number of element by oxygen
- Down a group, ionisation energy decreases, and size increases
=> Elements become more metallic and bonding to oxygen more ionic.
- Combination of Periodic and Group trends
=> change from ionic → polymer → small molecule
occurs later down the group as the Group Number increases.
Higher oxidation state oxides are more covalent, and more likely to contain double bonds. Even transition metal oxides, e.g. CrO3, Mn2O7, OsO4 have localised M=O bonds. Lower Steric crowding, since fewer O atoms needed for the higher oxidation state if M=O bonds used. Thus, Oxygen will stabilise high oxidation states (only Fluorine is better).
Why M=O bonds?
Thermodynamics – strong multiple bonds with small, highly-charged metals
Lower Steric crowding, since fewer O atoms needed for the higher oxidation state if M=O bonds used
- Therefore: Oxygen will stabilise high oxidation states (only Fluorine is better)
- Largely ionic oxides represent a vast array of extended solids.
There is also a structural classification that can be used if necessary. Down a group, ionisation energy decreases, and size increases.
- Elements become more metallic and bonding to oxygen more ionic.
Sulphur
Occurrence –
Mainly combined form, elemental, or H2S. Also pyrites, e.g. FeS2.
Elemental Form & Allotropes –
Allotropy most varied of all elements, along with Silicon. This shows a marked temperature dependence.
Extensive allotropy due to –S-S- catenation. The S-S bond is variable and flexible, as well as being very strong.
Sulphur in its elemental form typically forms cyclic S8 units. The most common is a yellow, orthorhombic α-form. This has D4d symmetry and very low thermal conductivity.
At about 100oC the α-form converts to β-S8, which has lower density. It shows the same ring structure, but they are packed in a more disordered fashion.
Heating to above 150oC and then cooling gives γ-S8 – more efficient packing and thus increased density.
It should be noted that solids of Sn (n = 6-20) can be formed. Also liquid sulphur has unusual properties. Gaseous Sn (n = 2→10) where n depends on temperature. Up to 600oC it is still S8, but above 720oC forms S2 (S=S) with structure as for O2 at room temperature.
Reactivity –
Sulphur is very reactive at slightly higher Temperature – presumably as this facilitates cleavage of the strong S-S bond.
Some typical reactions are:
- Heating with H2 to 100-200oC.
- Ignites in F2 to form SF6.
- Heat with Cl2 forms S2Cl2.
- Dissolves in liquid Br2 gives S2Br2 but this dissociates readily.
- In iodine, it behaves as a solvent, giving no compound.
Oxidation in O2 at rtp proceeds slowly to SO2. It will ignite in air at 250oC.
Sulphur forms a diverse range of compounds – covalent, coordinate, ionic and even metallic.
It can exist as a polyatomic cation (by oxidising in SbF3), e.g. [S8]2+, which is deep blue and cyclic. [S4]2+ also exists and has D4h symmetry, and is yellow. Paramagnetic Sn+ ions have also been found, but not much is known.
As a ligand, Sulphur can be terminal or bridging. It traditionally forms class-b (soft) ligands, indicating they are soft Lewis Bases. Qualitatively, this can be explained by Sulphur’s increased size meaning it has a deformable electron cloud and dπ orbitals available in bonding.
Range of Oxidation States –
Quite a range of oxidation states known, from -2 (H2S), -1 (S22-), 0 (elemental S), +4 (SO2) and +6 (sulphates).
Types of Compounds –
Sulphides
These are typically ores of metallic elements.
They are known to form a range of impure nonstoichiometric phases and thus are hard to make. Methods typically are:
- Direct combination
- Reduce sulphate with carbon
- Precipitate from aqueous solution with acid and H2S (or alkali as (NH4)2S).
- Saturation of alkali hydroxide with H2S to form MHS which then forms MS + H2O.
The solubility of MS compounds vary considerably. For example, Group 1&2 metals form highly ionic compounds which are thus highly soluble, although hydrolysis often leads to MSH + MOH. This is pH dependent. Conversely, Group 11 & 12 heavy metals form some of the least soluble compounds known.
As far as structures are concerned, these again can vary considerably although can be broadly characterised:
Ionic M2S (Group 1 metals) = antifluorite.
Ionic MS (Group 2 metals) = rock salt (also for less basic metals elsewhere in Periodic Table).
Later Transition Metals – covalency leads to lower coordination number or layer lattices, e.g. Zinc Blende and Wurzite.
Also the NiAs structure often appears, particularly for binary compounds of the 1st row TM’s.
Disulphides are also well characterised – consisting of Metal + discrete S2 units. For FeS2 there are two forms – pyrite and marcasite. The marcasite structure is similar to rutile, but not occupied by any other compounds. Pyrites are quite common and are similar to NaCl structure, except that the rod-shaped S-S unit replaces the Cl.
The sulphides show a vast range of physical properties and cannot be broadly described.
Anionic polysulphides also exist, and are similar to FeS2 but have the form MmSn.
Hydrides
Also known as sulphanes. H2S is the only stable one. It is widely found in nature, e.g. at volcanic or bacterial action. It is foul-smelling and very poisonous.
It can be formed by many reactions. One typical one would be FeS + HCl.
H2S shows far less H-bonding than water, and so its physical properties differ greatly. It is readily soluble in acid and alkaline. It also burns in air to form SO2 + H2O. It can also be used as a mild reducing agent.
Polysulphanes, H2Sn (n = 2-8 obtainable pure) consist of unbranched Sn chains – showing Sulphur’s tendency for catenation. As n increases, density, viscosity and boiling point increase. They are all yellow except n=1,2. They all readily oxidise and are thermodynamically unstable with respect to disproportionation:
H2Sn → H2S + [ (n-1)/8 ] S8.
Halides
The 7 fluorides differ from other halides of sulphur. They can show Oxidation States for Sulphur from 1 to 6, and Coordination Numbers from 1 to 6 also. It is rare to see such varied structural isomerism in simple molecular compounds, e.g. FSSF and SSF2. S2F2 is similar in structure to H2O2, O2F2 and H2S2. SSF2 however has sulphur in +4 and +2 oxidation states, although the S=S bond length here is similar to the S-S of FSSF.
SF2 is a fugitive species that dimerises by inserting into the S-F bond to give F3SSF. This is in stark contrast to SCl2 SH2.
SF4 is very reactive and used as a fluorinating agent. It is actually an amphoteric Lewis Acid-Base (donates & accepts). It hydrolyses to HF and SO2, and oxidises to OSF4 (this is slow in O2 at rtp).
Direct combination of sulphur and fluorine gives SF6, an octahedral molecule and illustrates sulphur being hexavalent. It is a highly stable molecule, especially when compared to the absence of SH4 and SH6, even though S-H and S-F are similar strengths. This is in fact due to the electronegativity of F favouring polar, 3c4e bonds as required. Also F-F is weaker than H-H. 3d orbitals on S also play a part, since S bears positive character and so these orbitals will contract, making them energetically and spatially more favourable for overlap with F orbitals.
SF6 is highly unreactive. It is odourless, tasteless, colourless, non-flammable, non-toxic and insoluble. It can be heated to 500oC without decomposing, and heating with metals yields no reaction. It is frequently used as an insulating gas, although there are concerns as it is a potent greenhouse gas. It does not react with hot steam either, though this is believed to be due to kinetic reasons. It will react with H2S to form S + HF. Boiling Na and Na in liq. NH3 reacts to form Na2S. Successfully oxidising SF6 to SF5OF (hypofluorite) gives a very reactive yellow compound.
A by-product of S+F reaction is S2F10 which is quite stable (not hydrolysed by water or dil. acid/alkali) and toxic. It disproportionates at 150oC by radical SF5.
A range of chlorides are also known. S + Cl2 gives S2Cl2, a toxic, golden-yellow liquid with a revolting smell. This reacts further with FeCl3 to give SCl2, which is cherry-red, foul-smelling, toxic and unstable when pure. Both these compounds hydrolyse easily to give a range of compounds: H2S, SO2, H2SO3, H2SO4.
Oxidising SCl2 yields O=SIVCl2 (thionyl chloride) which can be further oxidised to O2SVICl2. The oxohalides of Fluorides are more varied, including peroxofluoride compounds.
S can also catenate when bound to chlorine, forming SnCl2 compounds (n=1-8). This does not occur with F due to the electronegativity of F compared to Cl, and S-Cl is weaker.
Sulphur Bromides are poorly characterised, since SBr2 does not exist at room Temperature and S2Br2 readily dissociates into its elements.
Oxides
There are 13 proven oxides of sulphur. SO2 and SO3 are the most stable of them.
There are six SnO homocylic polysulphur monoxides. These are made by oxidising the Sn cycle using CF3C(O)OOH. S7O2 and S6O2 are also known.
S2O, S2O2 and SO are all thermally unstable acyclic oxides. SO is a fugitive species (unlike O2!)
SO2 can be made by heating H2S or pyrites in air. It is typically the starting point for making sulphuric acid. It is colourless and toxic, and soluble in water. Liquid SO2 is a non-aqueous solvent. It oxidises to SO3. It also behaves as a ligand to metals with 0/+1 oxidation states. It can adopt a variety of geometries – planar, pyramidal, bridging or side-on (9 modes in total).
SO3 is also part of sulphuric acid production. It is a liquid which reacts aggressively with most materials when anhydrous. It is planar as a gas (D3h), and reversible with a cyclic trimer S3O9. Its key reaction is to react vigorously with water to form H2SO4. It is also a Lewis Acid so forms a variety of adducts.
Higher oxides can be formed by combining SOx + O2 with electric discharge to form SO3+x polymeric compounds.
Oxoacids
There are many of these, all connected by a series of redox equilibria. Few of them can be obtained as a free acid – most as aq solution or as crystal salts.
H2SO4 is the most common, and in the anhydrous form is a dense viscous liquid. It is miscible in water in all proportions, which occurs extremely exothermically. It has an extremely high Dielectric constant and is highly conductive due to self-ionic dissociation.
It also forms salts (sulphates and hydrogen sulphates) with metals. SO42- is also a ligand (mono/bi/bridging).
Sulphur-Nitrogen Compounds
S4N4 is a tetrameric pseudocluster. It has unusual bonding (Lewis Model is insufficient to depict) and formal oxidation states here are not appropriate. There are many resonance hybrids. It is noted that S & N are diagonally related in the periodic table, so have similar electron charge density for similar coordination, as well as similar electronegativity, so this may explain this.
Binary SxNy compounds show little similarity to those of OxNy. NS is unstable. As mentioned, S4N4 is stable, and forms yellow-orange crystals with symmetry D2d. Kinetically it is stable, but thermodynamically endothermic, and in fact may detonate when struck or heated rapidly. This is due to stability of S and N2 bond strengths.
It is insoluble and unreactive towards water but undergoes base hydrolysis to give thiosulphates. Conc. alkali gives the sulphite. It is a useful synthetic intermediate for forming other S-N compounds.
S2N2 can be formed by depolymerising S4N4 over silver wool. It is an unstable cyclic dimmer, and spontaneously polymerises to (SN)x, polythioazyl, which has a planar chain polymer structure. This compound has a bronze metallic lustre. Its conductivity increases as Temperature decreases, and is a superconductor below 0.33K. It is stable in air even when heated, but explodes at 240oC.
There are also 6 further sulphur nitrides.
Sulphur Imides, S8-n(NH)n form as NH is isoelectronic with S, so it subrogates into the S8 ring. The structure is similar.
Sulphur-Nitrogen-Halogen Compounds N-S-X can also be formed, but their stability decreases with weight of halogen. The halogen attaches to S rather than N, unlike the imide.NSF and NSCl are the simplest, and are a noteworthy contrast to O=N-X. The most electronegative atom will bond to the least electronegative, as is shown. NSF is colourless, reactive and pungent. The most common compound of this series is S3N3Cl3, which forms yellow needles when Cl2 is added to S4N4.
Sulphur-Nitrogen-Oxygen Compounds are also known, e.g. S3N2O2 is a yellow solid which is easily hydrolysed in moist air, and oxidised by SO3.
Amides of sulphuric acid, e.g. H[H2NSO3] (sulphamic acid), are stable. Sulphamic acid forms a strongly H-bonded network (Zwitterion units), and dilute aq. solutions are stable at room temperature (they hydrolyse at higher T). Alkali metal salts are stable in alkali.
Formal replacement of OH in sulphuric acid yields sulphamide, (H2N)2SO2. This is colourless, crystalline, decomposes above 93oC and is soluble in water. It has a similar structure to sulphuric acid.
Other Group 16 – Selenium, Tellurium & Pollonium
Occurrence –
Se and Te are rare. They are usually found in association with S, and occasionally native. Main source is the anode slime from electrolytic refining of Copper.
Po is exceedingly unabundant due to radioactive decay. All 27 isotopes are radioactive, of which only 210Po can be found naturally. Po is obtained from Uranium decay (Marie Curie).
Elemental Form & Allotropes –
8 distinct forms of Selenium -, α, β, γ = Se8 rings. They differ in their packing, and are all red. Other rings have been synthesised. There is also a grey metallic hexagonal close packed form of helical polymeric chains, which is the thermodynamically most stable one. Also vitreous black Se – irregular structure of very large rings. It is brittle, opaque, and transforms to grey Se at 180oC. Research carried out due to photocell usage.
Tellurium has only one crystalline form – a network of spiral chains. The rapid decrease in allotropic complexity from S through to Te is notable.
Pollonium has a unique structure – it is the only element of simple cubic form.
Reactivity –
Down the Group, they get larger, have a lower EI, and are less electronegative. This would be expected. There is a trend to metallic conductivity too. Se & Te are semiconductors, and Po is a metal. There is also a tendency for cationic (basic) properties down the group, e.g. Se not attacked by dil HCl but Po dissolves readily and is oxidised to PoO4.
All the elements combine directly with most elements, though less readily with O & S. Most stable compounds are the selenides etc, especially M2- with Groups 1, 2 and Ln (+ other electronegative elements).
Oxidation States are typically +2, +4, +6. Se & Te will also catenate, but not nearly as much as S. As found in preceding Groups, multiple bonds get less stable down the Group also, e.g. CO2, CS2 stable, where CSe2 polymerises and CTe2 is unknown.
Again as with previous Groups, the element following the 3d Transition Series resists oxidation to Group Valency (SeVI). Also notable is the decrease in stability of H2M down the group. This is due to M(IV) being more stable relative to M(0) and M(VI) for Se, Te & Po, but not S.
Compounds of Se, Te and Po are potentially toxic. Se is needed in humans in trace amounts. Po is extremely toxic at all concentrations.
Polyatomic cations and anions are known. The cations form brightly coloured solutions when dissolved, as for S. They are less electropositive than the equivalent S. The anions are formed by reducing the element with alkali metal in liq. NH3. They are more stable as Mx2- than as the corresponding hydride.
Types of Compounds –
Selenides, Telenides & Polonides
Binary chalcogenides. Much non-stoichiometry, particularly with Transition Metals where electronegativity differences are minimal, therefore variable valency. Many can be considered metallic alloys.
Best synthesis is by direct combination under heat in the absence of air. Most will decompose in water or dil. acid to form H2Se or H2Te.
Polychalcogenides are less stable than polysulphides, and tend to oxidise in air.
Hydrides
H2Se can be made by direct combination but H2Te and H2Po cannot due to thermal instability. H2Po can only be made in trace amounts. H2Te and H2Se are colourless, foul-smelling and poisonous. They are soluble in water, but not to the extent of hydrogen sulphide (increasingly acidic solutions). They burn in air with a blue flame to give the dioxide. They also react with halogens readily and rapidly. Also true for aqueous solutions, to yield a precipitate of the elements.
Halides
Selenium forms no binary iodides, but Te and Po do, since more electropositive. There are numerous Cl and Br compounds known, particularly in Oxidation States of +1, +2, +4. For +6, there are only fluorides. SeF4 and TeF4 are known, but there are no lower fluorides except fugitive species.
The lower halides tend to form chains and polymerise, e.g. Te3Cl2 chain and TeI polymer. Se2Cl2 and Se2Br2 are highly coloured heavy liquids, with structures similar to S2F2. Several mixed species are also known. PoBr2 and PoCl2 are known, but doubt has been cast on the Te equivalents as they disproportionate rapidly, and the SeX2 are unknown.
All 12 tetrahalides are known except perhaps SeI4, and can be made by direct combination. They span the covalent-ionic border. SeF4 is a colourless reactive liquid which fumes in air, and crystallises to a white hygroscopic solid. It is a useful fluorinating agent, with structure as SF4. TeF4 is similar but much more reactive, e.g. readily fluorinates silica. Tetrahalides tend to be good electrical conductors due to self-ionic dissociation. Detailed structures of PoX4 are not known.
The only hexahalides known are SeF6 and TeF6, and they form colourless gases from direct combination. They are both octahedral are relatively inert. TeF6 hydrolyses after one day (cf. SF6).
Numerous oxohalides are also known. SeOF2 and SeOCl2 are colourless fuming, volatile liquids. SeOBr2 is less stable orange solid that decomposes in air at 50oC. They can be made by combining SeO2 with tetrahalide, and have structures as for OSF2. SeOF2 is an aggressive reagent that attacks glass and silicon slowly, and has a structure of linked OF bridges to form layers (unlike discrete SOF2). It is also a useful solvent which conducts.
Oxohalides of SeVI only form with F, and tend towards the hypofluorite F5SeOF, or peroxide-type equivalent.
Oxides
SeO and TeO have transient existence in flames. PoO has been obtained as a black easily oxidised solid. Dioxides of all 3 elements are however well established.
SeO2 is a white solid which melts to a yellow liquid. It is very soluble to give selenous acid, H2SeO3. It is thermodynamically less stable than SO2 and TeO2 and can be easily reduced to elements by ammonia. It is a useful oxidising agent in organic chemistry. The solid implies a polymeric structure of corner linked pyramids of [SeO3].
TeO2 is dimorphic, both forms involve the [TeO4] unit – one is rutile-like while the other is orthorhombic. TeO2 is much less volatile than SeO2, and is not very soluble. It is amphoteric.
PoO2 is a yellow fcc fluorite lattice, and sublimes. It is amphoteric, but appreciably more basic than TeO2.
The Group 16 Oxides favour higher coordination down the group:
- SO2 – molecule
- SeO2 – chain polymer
- TeO2 – layer or 3D.
- PoO2 – 3D fluorite.
SeO3 is unstable wrt dioxide. It is a white hygroscopic solid. TeO3 again has 2 modifications, one of which (grey) is less reactive. Both are not attacked by water, but the yellow/orange form is a powerful oxidising agent when heated. PoO3 is uncharacterised.
Hydroxides and Oxoacids
The rich oxoacid chemistry of S is not paralleled. Only compounds of +4 & +6 are known. Selenous acid, O=Se(OH)2 and the Te equivalent (less stable) are white solids. PoO(OH)2 is a pale yellow precipitate from hydrated PoO2 and is appreciably acidic.
In +6 oxidation state, Se & Te differ greatly. H2SeO4 is a viscous liquid which has properties similar to sulphuric acid, which Te(OH)6 is a white octahedral solid. Telerates are not isomorphous with sulphates and selenates.
There are also numerous peroxoacid and thioacid derivatives of Se & Te. The increased basicity of heavier Group 16 leads to more oxoacid salts, particularly for Po.
Other Compounds
Se4N4 – red covalent molecular species. There are other mixed compounds of Se8 / S8 rings. They are structurally equivalent.
PoS – black precipitate of H2S + acidified Po.
Se-N and Te-N bonds known but not as varied as S-N. Se4N4 is an orange shock sensitive crystalline compound which decomposes violently at 160oC. It is D4d as for S4N4. It is also thermochromic (yellow/orange at -200oC and red at 100oC). Te3N4 is lemon-yellow, violently explosive, and its structure is unknown.
GROUP 17
Fluorine, Chlorine, Bromine, Iodine, Astatine
Occurrence –
Reactive, therefore not free state. Widespread in abundance as X-. Iodine also occurs as iodate. Astatine is fugitive, radioactive and can hardly be said to exist in nature (partakes in radioactive decay of other elements). It is the rarest naturally occurring terrestrial element.
Elemental Form & Allotropes –
Volatile diatomics, and colour increases with mass – yellow → green → dark red → lustrous black.
There are few isotopes (typified by odd atomic number).
In the solid state molecules align to give a layer lattice which is orthorhombic for all but F2. They are all poor conductors, though iodine is a 2D semiconductor (depends on plane) and compressing it makes it metallic.
Extraction:
F2: by electrolysis of KHF2
Cl2: by electrolysis of NaCl/H2O → NaOH + Cl2
Br2 & I2: by Cl2 oxidation of Br-, I-
Colours of elements due to π*→σ* excitation.
For I2 transition is perturbed in different solvents.
In non-aqueous solvents can get Hal2+ species
Order of stability I>Br>Cl
I2 / conc. H2SO4 → I2+, blue paramagnetic, r(I-I) < than I2
Br2+ [Sb3F16]- (Cl2+ analogue unstable at 20oC)
Unique Aspects of Fluorine
- Due to its Position in Periodic Table and Small Size
- Unexpectedly Low Bond Dissociation Energy of F2 w.r.t. other Hal2
- Lone pair repulsion most significant for F2
- pπ→dπ bonding may supplement bond strength for Cl2, Br2, I2
- Interelectronic repulsion greatest for small halogen (this is by far and away the most important term, caused by the exceptionally small size of F)
- Strong Bonds to Other Elements, where non-bonding pair repulsion minimal, if π-bonding to empty orbitals on E occurs.
- Valence Expansion of Combined Elements e.g. IF7, SF6, UF6
- small size → space for high C.N. highly oxidizing → maximum oxidation state.
- No Expansion of Fluorine's Octet! e.g. no F analogues of ClO3- or Cl2O7
- Note in e.g. S-F-S bonds in S2F10 S-F bridge bond is weaker than S-F terminal bond
- High Electronegativity → Strong Hydrogen Bonding, e.g. structure of (HF)x → Large Inductive Effect [ e.g. acid strength of HFSO3 ]
- Small Size of F- → High Lattice Energies / Hydration Energies
Reactivity –
- Group 17 (VIIA) Electronic Configurations [N.G.] ns2np5
- Accessible oxidation states -1→+7,
- Obeys the Octet Rule → Lone pairs play important role in chemistry.
- Plenty of aqueous redox chemistry.
- No simple monatomic cations in aqueous solution
Electron Affinity is at a maximum for Cl > F > Br > I. Enthalpy of Dissociation is similar as Cl2 > Br2 > I2 = F2. Just as N-N is weaker than P-P and O-O weaker than S-S, this trend is due to partial pd hybridisation giving some double bond character to P-P, S-S and Cl-Cl.
F-F is particularly weak due to less overlap of bonding orbitals, appreciable internuclear repulsion and large e-e repulsion of lone pairs.
F is the most reactive of all elements. It combines with everything except He, Ar & Ne. Often direct combination is vigorous and explosive. There is a tendency for F2 to form F- in solution, and this tendency is much greater than for the other halogens. F2 is also an extremely strong oxidising agent, hence high oxidation state compounds for elements with which it reacts, e.g. IF7 and PtF6.
As with all 1st row, F differs from other X due to a) small, b) electrons tightly held, and c) no low lying d-orbitals. Ionisation energy for F is thus much greater than for other X, so there are no positive oxidation state and F is exclusively univalent. Note that technically lone pairs allow F to act as a Lewis Base for Coordination Number > 1.
The heavier halogens are still reactive, in the order Cl > Br > I. In general, reaction of X2 with M-M, M-H, or M-C results in M-X. Solubility of halogens varies greatly depending on solvent, and solvolysis or halogenation may occur. Colour usually indicates solubility.
Types of Compounds –
Hydrogen Halides
HF is a colourless volatile liquid and oligomeric H-bonded gas. Other H-X are colourless diatomic gases. This is due to lack of H-bonding compared to HF. HF is less viscous than water, which indicates no 3D structure of H-bonds.
Anyhdrous HX are versatile and vigorous reagents for halogenation of most compounds (dependent on thermodynamics, catalysis, other kinetic factors). There are numerous hydrated forms for all HX except HI.
HF is miscible in water in all proportions, with many H-bonds. Such H-bonds are very relevant as other aqueous hydrohalic acids are extremely strong while hydrofluoric acid is very weak. O-H------X for X = Cl, Br, I is very long and weak, so a strong acid.
HF as a solvent has a high Dielectric constant, low viscosity, long liquid range and so a good solvent. However anhydrous HF attacks glass. Self-ionic dissociation occurs so that it conducts. Most fluorides dissolve to give F-, but a few don’t, e.g. XeF2, SF6, MF6 (M = Mo, W, U, Re, Os). Also, liquid HF is used to dissolve biological molecules, since it has little chemical consequences, while prevent dehydration.
When not fluorides, solvolysis usually occurs (evolution of HX). Other hydrogen halides are less useful solvents due to physical properties mentioned.
Halides
Wide range of stoichiometries, structure types and properties. No unified classification. As expected, fluorides tend to be ionic structures while larger more polarisable other halides tend to adopt layer lattice or chain structures.
Born-Haber cycle for the synthesis of a halide compound:
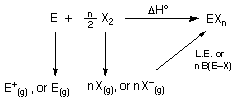
In general, fluorine stabilises high oxidation state compounds.
Explanation of trends:
1. Weak F-F bond (also kinetic argument).
2. Strong B(E-F) bonds, (F > Cl > Br > I).
3. High electron affinity, cf. ΔHf[X-(g)].
4. High lattice energy, small F- ion (F>Cl>Br>I), Kapustinskii approximation.
Low to intermediate oxidation states. EF2 unstable with respect to disproportionation (e.g. SF2).
- Covalency increases with oxidation state.
- Covalency increases down the series F → I.
- Lower oxidation state tends to be formed with Br and I.
- High oxidation state fluorides tend to be molecular.
Fluorides
Stoichiometry from C4F to IF7. Synthesis can be effected by halide metathesis in aqueous solution, assuming hydrolysis poses no problem. Otherwise, anhydrous HF is used. F2 is usually avoided due to cost and difficulty of handling. Can also use halogen fluorides, as well as suitable fluorides (hard = MnF3, PbF4, soft = HgF2, CaF2, SbF5).
Fluorination reactions can be categorised as metathesis, oxidation or substitution. Occasionally reductive fluorination is used.
Chlorides, Bromides & Iodides
Similar set of preparatory routes. Can be hampered by hydrolytic disproportionation to X2 or hydrolysis as before.
Groups 1, 2,3 + Ln/An form halides that are predominantly ionic. The covalent-ionic transition is roughly around Group 15/16. Bond types tend to show a gradual change though. Type of bonding can usually be elucidated by solubility or reactivity.
Interhalogen Compounds
Halogens combine exothermically with each other to form XY, XY3, XY5 & XY7 (X = heavier halogen). A few ternary compounds are also known. For the hexatomic series there are only fluorides, and the only octatomic compound is IF7. All interhalogens are diamagnetic. Chemically similar reactivity is shown by XCN, XSCN & XN3 imply that CN, SCN & N3 are pseudohalogen compounds.
XY
All six are known. Variable stability e.g. ClF stable, BrCl readily dissociates reversibly and BrF & IF disproportionate rapidly and irreversibly. Generally, properties are intermediate between those of parent halogens. They are often aggressively reactive, so hard to characterise. Structures vary. Reactions are typically of the type:
- Halogenation
- Donor-accept interactions
- Use as solvent systems
Reactions frequently parallel parent halogens. ClF is more reactive ICl and IBr. They all react with most metals.
XY3
ClF3, BrF3, IF3 and ICl3. All prepared by direct reaction. Controlled to avoid mixtures. IF3 not stable above -30oC (forms IF5). ICl3 dimerises and is a bright yellow solid, which readily dissociates to ICl + Cl2. ClF3 & BrF3 are volatile molecular liquids with an unusual T-shaped structure (C2v). I2Cl6 is planar.
ClF3 is one of the most reactive compounds known. It spontaneously ignites asbestos, wood & other building materials and is often used in bombs. It explodes with H2O and organic substances. Moderation can be achieved by dilution with an inert gas. Only suitable containers are mild steel, copper & particularly Nickel. Pure ClF3 cannot attack pyrex or quartz but traces of HF will. ClF3 converts chlorides and oxides to fluorides. Manufactured on a relatively large scale considering risk since it is used in nuclear fuel processing to produce UF6 gas (separates from Pu). ClF3 is also a fluoride ion donor or acceptor.
BrF3 is still very reactive, but not as reactive as ClF3. It is also used to make UF6. The trend for reactivity amongst interhalogens is typically Cl > Br > I and XF5 > XF3 > XF.
Other interhalogens are all extremely vigorous fluorinating agents. Most stable interhalogens thermodynamically are ClF3, BrF3, IF5. XF5 is square pyramidal (C4v). IF7 is a pentagonal bipyramid (D5h).
Polyhalide Anions, XY2n- (n=1-4)
Halide + interhalogen, or halide transfer generates these. There are ternary forms also, but the total number of atoms in the anion is always odd. There are numerous of these anions for the iodides.
They are stabilised by large cations such as Cs+ or PCl4+. For a given cation, the stability increases by symmetric polyhalide ion and larger central atom. The sequence is thus:
I3- > IBr2- > ICl2- > I2Br- > Br3- etc.
Triatomic units are usually linear. Pentatomic are usually square planar, while heptatomic are non-octahedral even though there are 14 valence electrons. IF8- is also known by Raman Spectroscopy only.
Iodine can catenate quite effectively, hence the number of polyiodides, which can be as large as I9-.
Polyhalonium Cations, XY2n+
These are halide ion donors. The colour depends very much on the weight – those with F are colourless, but ICl2+ is red/orange and I2Cl+ is brown/black. Their structures are as expected, e.g. structures of triatomics are bent.
Halogen cations also form, e.g. iodine dissolves in strong oxidising solvents to give bright blue paramagnetic I2+. Br2+ and Cl2+ also forms as expected, since the ionisation energy of O2+ (which is known to form) is higher. However Cl2+ is not stable enough to characterise.
Formation of these halogen cations usually requires SbF5 or a similar compound. Raman Spectra are used to characterise the numerous halogen cations. The triatomics are non-linear as mentioned, and are isostructural with 20e species. I3- on the other hand, with 22e, is linear.
Oxides
Show diversity amongst the halogens. Varying relative strengths of O-X bonds due to varying electronegativity. There is also as a consequence varying redox properties.
The OF compounds have been discussed under Oxygen, but there are also 25 other binary halogen oxides.
Chlorine Oxides
These show instability. They are all endothermic (thus cannot be prepared from Cl2 + O2). They are much studied due to the atmospheric consequences.
Cl2O – brown gas. Explodes when heated / sparked. Very soluble in water as HOCl. It is used to make hypochlorites, and is also a bleach. Preparation is usually from Cl2 + HgO → Cl2O.
Cl2O3 – dark brown solid. Explodes below 0oC.
ClO2 – yellow paramagnetic gas. Explodes at -40oC. It is stable to dimerisation just like NO, which is again due to delocalisation of the odd electron. Used for bleaching and water treatment. Can be made by reducing NaClO3. It is a strong oxidising agent to both organic and inorganic materials, and decomposes slowly in the dark. Light yields ClO + O which has many consequential photolysis reactions – relevant in the stratosphere. It also hydrolyses to HCl + HClO3, and in alkali forms the chlorite and chlorate.
Cl2O4 – pale yellow liquid ClOClO3. Decomposes at room temperature.
Cl2O6 – dark red liquid in equilibrium with ClO3. Decomposes. The dimer does not show up as Cl bridges (lack of paramagnetism), but is likely somewhere between [ClO2]+[ClO4]- and O-bridging. It hydrolyses to form HOClO2 + HClO4 (perchloric and chloric acids).
Cl2O7 – colourless oily liquid. This is less reactive than the lower oxides, but still shock-sensitive compound.
There are also the short-lived radicals ClO, ClOO & ClO4.
Bromine Oxides
Less numerous or as characterised as those of Cl.
Br2O is moderately stable at -60oC and is an oxidising agent.
BrO2 can be formed by low temperature ozonolysis of Br2. It is actually BrOBrO3 with BrI and BrVII. It decomposes violently at 0oC.
Br2O3 detonates at 0oC as well. It is formally the anhydride of hypobromous and bromic acids.
There are again also some unstable monomeric radicals.
Iodine Oxides
These are the most stable oxides of the halogens.
I2O5 – O2IOIO3 formed from the joining of 2 x pyramidal IO3 by a bridging O. It is the dehydrate of iodic acid (via dry air at 200oC). It is a white hygroscopic crystalline solid which is thermodynamically stable. It is one of the few chemicals that oxidises CO rapidly and completely to carbon dioxide.
I4O9 & I2O4 are less stable. There are again short-lived radicals IO, IO2, IO3. The higher oxides are much more stable for I than they are for Cl and Br. Consequently, the IO radical is far less stable than that of Cl and Br.
Oxoacids
There is a vast range of aqueous solution chemistry. They are all to some extent soluble, but extensive disproportionate and/or redox may occur with the solvent.
There is only one oxoacid of fluorine. This is because the ½ F2 / F- couple has a high standard reduction potential of +2.866V, therefore it reacts with water at all pH. Also, the limitation comes about by fluorine’s resistance to oxidation states higher than +1, leaving just HOF (hypofluorous acid).
The oxoacids are numerous for the other halogens. There is a decrease in oxidising power down the group (due to Electrode Potentials). For XOnm- salts, an increase in pH causes Eo to decrease dramatically. Thus Cl2 / Br2 much more stable in acid wrt disproportionation.
Hypohalous Acids = HOX. HOF is hard to make as it reacts with HF or H2O involved in the reaction. It is non-linear and unstable, decomposing to HF + O2.
HOCl is far more well-known. It is used for bleaching and sterilising, and is more stable than other HOX. They are all highly reactive and relatively unstable though, but known in aqueous solution.
The hypohalites formed from them are amongst the strongest common oxidising agents.
Halous Acids = HOXO. HOClO is in fact the least stable oxoacid of chlorine. The Br and I equivalents are even less stable. The anion chlorites are more stable, but there are no bromites or iodites. ClO2- is a non-linear anion used to scrub off industrial gases such as NOx and H2S.
Halic Acids = HOXO2. NaClO3 is highly desirable and made on a very large scale by the electrolysis of brine. Chloric acid and bromic are both strong acids in aq, but iodic acid is weak. HOClO2 is fairly stable unless heated.
The halates are pyramidal anions. Their oxidising power is in the order ClO3- = BrO3- > IO3-, but reaction rates are IO3- > BrO3- > ClO3-. These are both dependent on pH.
Perhalic Acid = HXO4.
The most stable compounds of Chlorine are those in which the element is in either its highest (VII) or lowest (-I) Oxidation States. Thus perchlorates are the most stable oxo compounds of chlorine. When heated they tend to decompose by loss of O2. They are not notable oxidising agents at room temperature but when heated they become vigorous and violent. Pure HClO4 is a colourless, mobile shock-sensitive liquid with at least 6 hydrates. There is some H-bonding. The anhydrated form is a powerful oxidising agent, and reacts explosively with most organic materials. The perchlorates are known for most metals, and very stable for those with alkali metals.
Interestingly, the perbromates and perbromic acid were originally thought not to exist. Synthesis of KBrO4 has since been affected, and it has the structure isomorphous with KClO4 (tetrahedral perhalate ion). The difficult in preparation is that only the strongest oxidants can convert bromates to perbromates. The perbromates are, like the perchlorates, fairly unreactive for oxohalogen compounds.
Periodates are connected by a complex series of equilibria involving deprotonation and aggregation of the parent acid H5IO6. They are thermodynamically potent and kinetically facile oxidants. The oxidation potential is greatest in acid, where it can convert MnII to MnVIIO4-. It is often used in organic chemistry to cleave diols to diketones. Periodates can also form numerous complexes with transition metals as bidentate chelating ligands.
Other Compounds
Halogen Oxide Fluoride compounds (FnXOm) compounds are known with a range of stabilities and they resemble the halogen fluorides.
There are also numerous halogen derivatives of oxoacids, e.g. XOClO3 and XONO2 (where the H atom of an oxoacid has been replaced by a halogen atom).
Astatine Chemistry
As mentioned, all the isotopes of astatine are intensely radioactive with very short half-lives. No weighable amounts can be prepared and bulk properties are not known. Studies on it have shown that it is the only halogen with an oxidation state between 0 and V that is stable wrt disproportionation. At(0) will react with X2 to form AtX compounds, while halide ions yield AtX2-.
Group 18
The Noble Gases –
Occurrence –
Noble gases in the atmosphere. Helium is the second most abundant element in the universe, but is too light to be retained by the atmosphere during planet formation. The noble gases constitute 1% of the atmosphere, with most of that being Argon. Smaller concentrations are trapped in igneous rocks.
Elemental Form & Allotropes –
There are several isotopes of Kr and Xe, but only realistically one each of Ar and He. They all have stable electronic configurations (full p shell for all except He – 1s2). They are colourless, odourless, tasteless, monatomic and non-polar, hence their properties vary regularly with atomic number. Interatomic interactions only via weak Van der Waals.
Helium has unusual phase properties (superfluidity) which has been attributed to unusual/weak dispersion forces on the quantum level. Most noble gases, especially He, will diffuse through most materials, including rubber and PVC. Helium will even diffuse through glass.
Reactivity –
Xenon’s ionisation energy is in fact lower than that for O and F (EI drops down the group), and this explains why Xe is reactive for a noble gas.
In fact Kr, Xe and Rn will form fluorides, but Radon is very radioactive. Lighter noble gas compounds are merely transient unstable species.
Clathrates – noble gas + quinol, water or 1,4-C6H4(OH)2. Molecules of gas are trapped in cavities of crystals of quinol (requires pressure to form). This is a means of storing noble gases.
Xenon Compounds
Oxidation States +2 through to +8. There is also a variety of stereochemistries. There are 3 fluorides: XeF2, XeF4 and XeF6. These are formed by direct reaction under controlled conditions.
XeF2 – white crystalline solid of parallel linear XeF2 units. It sublimes, and is a mild fluorinating agent. It dissolves in water, where it is stable unless base is present:
2XeF2 + 2H2O → 2Xe + 4HF + O2.
Aqueous solutions are powerful oxidising agents.
XeF4 – also white, crystalline, sublimable solid. It is square planar as a gas and solid. It has similar properties to XeF2, but is a stronger fluorinating agent. It is also hydrolysed instantly by water, giving a variety of products, one of which is XeO3 (risk as highly explosive, therefore must exclude H2O).
XeF6 – formed by prolonged heating of Xe and F2 under pressure. It is a crystalline colourless solid, although more volatile than XeF2 and XeF4. It is yellow as a liquid and gas. It is more reactive, and so a stronger oxidising and fluorinating agent. Hydrolysis will occur with great vigour, and again XeO3 is produced, even in glass. It has proven difficult to ascertain structure for this reason, but it is not a regular octahedron, but a non-rigid distorted one. It has multiple forms as a solid and comprises ions.
Other halides and odd-valent fluorides are not stable. The binary fluorides can be used to make other compounds, by four methods:
- Reaction with F- acceptors. Typically XeF2 used, e.g. reaction with pentafluorides of P, As, Sb, I to form salts of the type [XeF]+[MF6]- and related. XeF+ is an excellent Lewis Acid, and this has been used to make Xe-N bonds.
- Reaction with F- donors. Only with XeF6. It reacts with alkali metal fluorides to form MXeF7 (M = Rb, Cs) and M2XeF8 (M = Group 1, not Li). These lose XeF6 under heat and stability increases with weight of metal.
- F/H metathesis between XeF2 and anhydrous acid.
XeF2 + nHL → F2-nXeLn + nHF (n=1,2). L = large oxoacids. Many of the compounds formed are unstable.
- Hydrolysis & related reactions. Form oxohalides and oxides. XeOF4 = colourless volatile liquid. Square pyramidal structure formed by hydrolysing XeF6. This can be further hydrolysed to XeO2F2 and then XeO3. The reaction is difficult to control.
XeO3 – controlled formation by using a current of dry N2 to sweep XeF6 vapour into water. XeO3 is colourless, and the aqueous solution is xenic acid. It is a dangerous explosive comparable to TNT. XeO3 (aq) is an extremely strong oxidising agent. Salts of XeO42- are not stable, as they immediately and slowly disproportionate to XeVIII (perxenate – XeO64-) and Xe0.
Xe can bond to other atoms besides O and F, e.g. replacing F with –N(SO2F)2. These are less stable and even less Xe-C bonds have been characterised. Recently and Xe-Xe bond was formed, and at 308.7pm is the longest element-element bond known.
Other Noble Gases
No stable compounds of He, Ne or Ar.
Rn apparently forms a difluoride, but due to radioactivity cannot be characterised.
Kr has some chemistry, but far less extensive than Xe. KrF2 is stable, but other fluorides are not. This is a stronger fluorinating agent than XeF2, but rapidly decomposes in water. There are also some cationic species. All other compounds that are known for Xe are unstable for Kr.
I am happy for them to be reproduced, but please include credit to this website if you're putting them somewhere public please!