Bioinorganic Chemistry
Mostly adapted from Prof Armstrong's Lecture Notes, with a few Tutorial Essays thrown in.
GENERAL BIOINORGANIC NOTES
Entatic State
How proteins “control” metal ions. Rigid protein structures (e.g. β-sheets) can confer strain on the metal, forcing it to adopt geometry of the transition state for the reaction it undergoes.
“Entasis” is the Greek word for stretched, which gives us the clue that its effect is structural. It in fact relates to distortion of coordination geometries around the metal ion(s) in a protein, and has been proven to exist by a variety of structural studies, namely EPR and Mossbauer Spectroscopy. It can also be elucidated from general redox properties.
It occurs because of and is related to the catalytic efficiency of the enzyme, and that the protein can fold to generate special stereochemistries which help the metal to adopt a geometry closer to that in the Transition State of the reaction.
It is seen in many enzymes. It has been most studied in “blue” Copper centres, where Cu(I) is seen to not adopt the expected perfect Tetrahedral geometry, and Cu(II) does not adopt a perfect square planar arrangement either. These blue Cu centres are found in a variety of electron transfer proteins and oxidases, such as azurin, plastocyanin and cytochrome c oxidase. The purpose is to enforce minimal difference between Cu(II) and Cu(I) geometries, i.e. the active site is held in an entatic state to minimize the reorganization energy. This allows for extremely fast electron transfer.
Other examples of its presence are seen around zinc in Carbonic Anhydrase and carboxypeptidase (distorted tetrahedra), and also iron in ferrodoxins. It is not easily modelled with simple ligands - no pre-organised strain.
Methods of Analysis of Metals in Biology
X-ray Diffraction - protein structure and position of Metals (and separations if more than one). Poor at distinguishing metal atoms and diatomic ligands are hard to see. For this, use IR Spectroscopy.
EPR Spectroscopy - very useful for studying metal ions. Need them to be paramagnetic (odd electron) metals (S = n/2). Process is similar to NMR. Gives electron configuration information, and also shows any magnetic coupling.
Mossbauer Spectroscopy - good for Iron coordination chemistry generally. A 57Co source decays by electron capture to give an excited state 57Fe**. This then decays either straight to ground state (9%) or to a lower excited state (57Fe* - 91%). The energy gap between the ground and 1st excited is ∆E, and resonance is achieved by moving the Co source away from the 57Fe in the sample under test (57Fe is 2% abundant).
Cyclic Voltammetry - this can be used to study redox active centres, e.g. blue Cu centres.
Iron
Porphyrin Macrocycles

These are used in Fe and Co (B12).
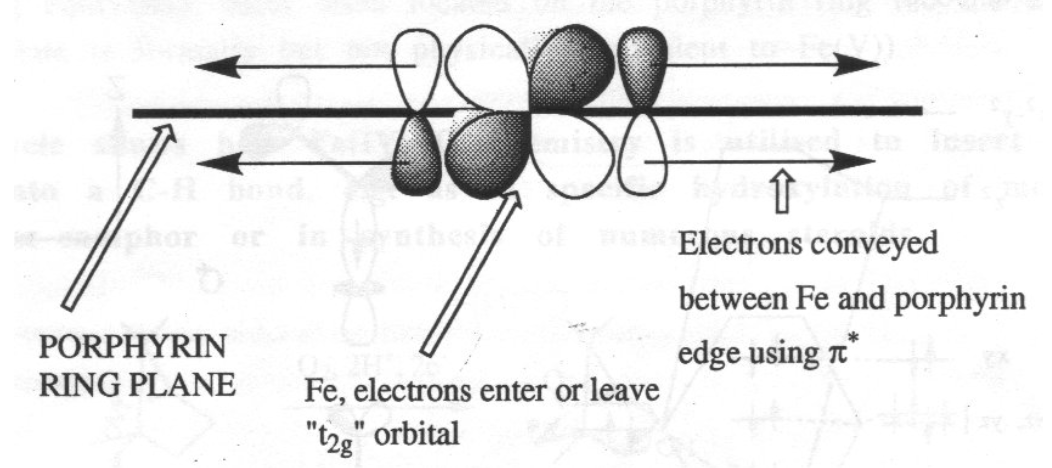
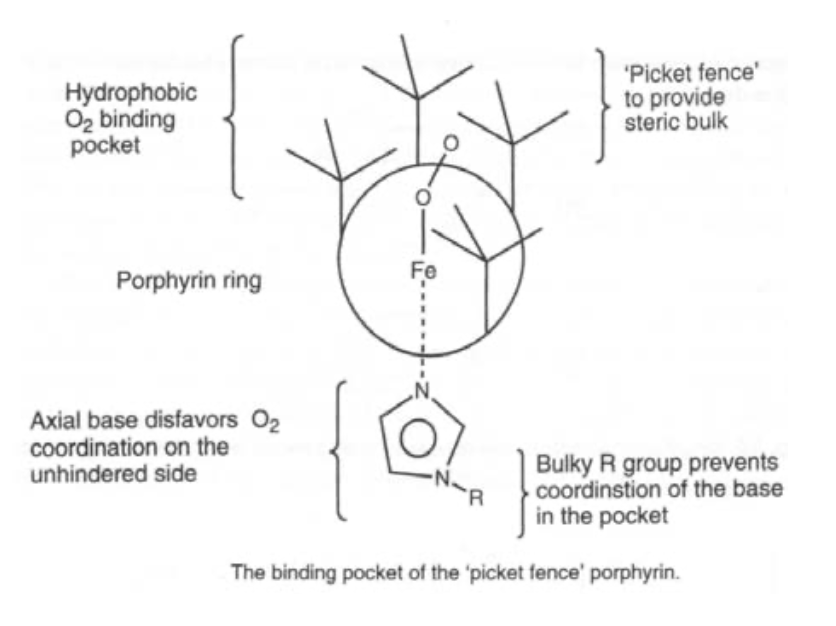
Note the delocalised π-system, and also that there is still the potential for two extra axial ligands to bind above and below the ring. This can carry Fe(II), (III) or (IV), although the latter is as FeIV=O (in biological systems only).
They find roles in electron transfer, ligand binding and catalysis.
Electron Transfer
Fe(III) + e- <=> Fe(II)
Eo ranges from +0.4 to -0.4 V depending on the ligands and the environment. Rates are determined by Marcus Theory - particularly the driving force, reorganisation energy and electronic coupling.
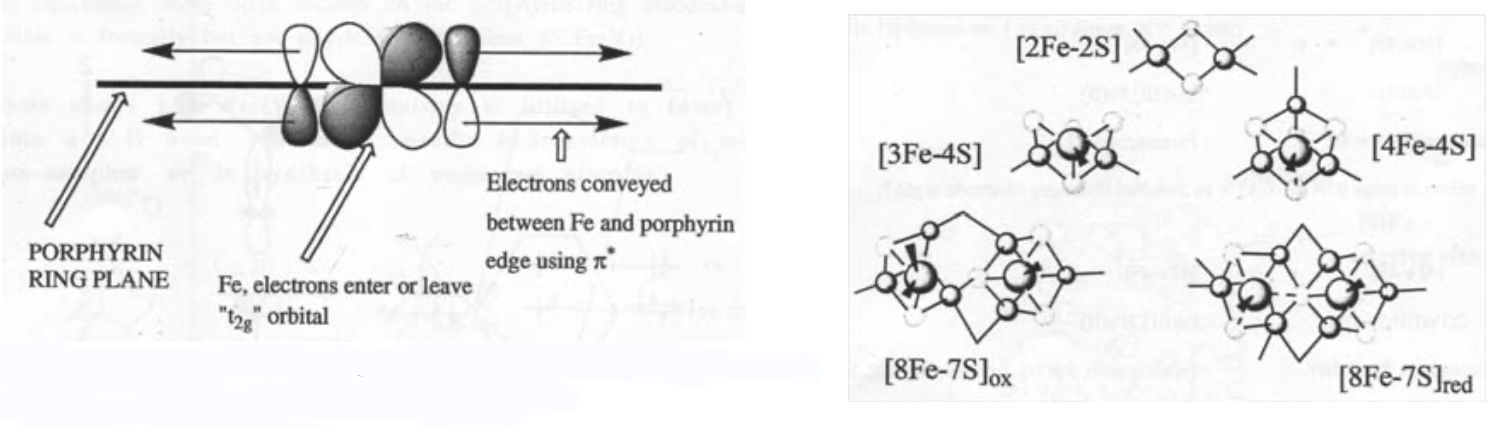
The heme ring ensures that the iron is always low spin, which accelerates the reaction greatly. It also acts as an electron antenna:
Cytochrome c is the classic example -
It occupies a position in chain of different cytochromes, and the whole process catalyses the formation of ATP from ADP:
NAD+ --> Flavoprotein --> Cyt b --> Cyt c1 --> Cyt c Æ Cyt c oxidase --> O2
It has Iron coordinated by a heme + two strong field ligands (Histidine and Methionine), so the iron is in its low spin configuration. This is advantageous for electron transfer as t2g6 <--> t2g5 (outer sphere) exchange will be far easier than any involving antibonding eg orbitals (the latter would require more distortion of the coordination sphere according to Marcus Theory, which expends energy and therefore slows the reaction).
Analysis of the crystal structure of cytochrome c containing both Fe(III) and Fe(II) has been accomplished. The structures are almost identical, which is further indication that the process is very fast, as a lack of change in conformation is ideal according to the Franck-Condon Principle (that is that electron transfer must occur between complexes of the same energy). This means that there is no need to adjust bond lengths between the two forms so that their energies are equivalent, i.e. that the free energy of reorganisation is very small. This results in very fast reactions according to Marcus Theory.
The structure also indicates that the iron is fully coordinated. This shows that the protein’s sole role is for electron transfer - no other reactions occur at its active site. This also renders it immune to other complexing ligands that may be toxic, e.g. cyanide, which deactivates cytochrome a. Another advantageous property is that the reaction is very clean. There are no other side-reactions and no harmful by-products or hazardous O-intermediates.
The mechanism by which the enzyme moves between the reductase (Cyt c1) and the oxidase (Cyt c oxidase) is not well understood. It is believed that when docking with the reductase, an electron is donated which is passed to the heme ring by π-electron cloud overlap, which gives rise to an ion pair. The fact that this is in a hydrophobic environment means that this is a very high energy intermediate, and this drives a conformational change that allows the electron to be shuttled between parallel residues, and compacts the protein and move to the oxidase.
Reversible Ligand Binding
Oxygen carrier. O2 binds end-on, and bent. It is also diamagnetic (i.e. the spins have been rearranged). Pauling explained this by considering singlet O2 binding to low spin Fe(II), such that the occupied π* on O2 acts as a σ-donor, and the empty π* acts as a π-acceptor. This gives the bent configuration.
Simple iron porphyrins do not show reversible ligand binding, as steric hindrance is required to stop oxidation.

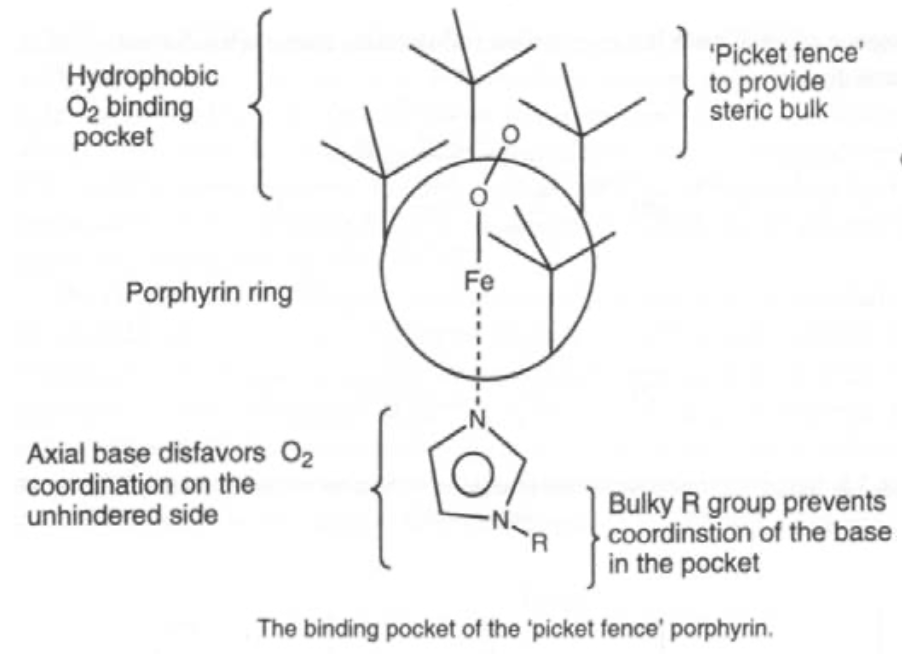
Clusters
Iron-Sulphur Clusters
Fe(III) and Fe(II) are connected by S2-. It is thus a mixed-valence compound.
Fe is usually found in tetrahedral geometry, and is high spin.
The clusters coordinate to proteins by thiolate ligands (Cysteine) to the Fe.
The most common ones are:
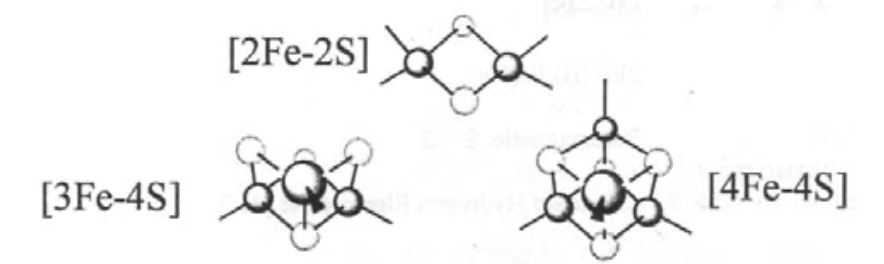
Super-clusters are also known, e.g. 8Fe-7S in nitrogenase.
The primary uses of these is in electron transfer, particularly for systems lying at the reducing end of process, i.e. e-transfer to/from enzymes that catalyse NADH/NAD+ and H+/H2 processes, e.g. Cytochrome P450.
Detection of the clusters is achieved by EPR or Mossbauer Spectroscopy. This gives information about Oxidation States and any magnetic interactions between Fe (ferromagnetic or antiferromagnetic coupling).
It is possible to make synthetic analogues under anaerobic and non-aqueous conditions, by using thiol ligands.
Biological Examples:
Ferredoxins - small proteins (MW ~ 12000), which carry ~2 clusters. They are essentially electron-carriers. Typically these are [3Fe-4S] and [4Fe-4S].
Hydrogenase - large protein (MW ~ 94000), which arrange the clusters in a line (wire-like) near the active site (NiFe). Thus sets up a chain of electron carriers.
Clusters are unstable with respect to Fe-O coordination (i.e. oxidation). They can be reformed by reduction though. These types of reaction are the basis by which a cell senses concentrations of Fe or O2 (relevant to protein production). Thus, FeS clusters transform in the presence of O2 or Fe which then alters the shape of the protein, and this then affects the binding of RNA/DNA.
Aconitase - acid-base catalysis
This catalyses the isomerisation of citrate to iso-citrate in the Citric Acid cycle, by moving the OH between C atoms. This isomerisation does not require Co-B12.
It uses a [4Fe-4S] cluster, and works by coordinating the substrate, polarising the bond, and then rearranging, all at a specific iron site. This shows the role of Fe(III) as a strong Lewis Acid. Under aerobic conditions, the enzyme is inactive as [3Fe-4S]. On reduction, it forms the cubane cluster, and is active.

Nitrogenase
N2 + 8H+ + 8e- + 32ATP --> 2NH3 + H2 + 32ADP + 32HPO42-
The problem with this process is the energetics - there is a large activation energy to reach the LUMO and N2 has a triple bond.
Nitrogenase achieves this, but requires a lot of ATP to do it. It is in fact a 2-protein enzyme. The smaller part has a 4Fe-4S cluster which binds to ATP. The larger part is a dimer with 2 super- clusters - [8Fe-7S] (“P cluster”) and [Mo7Fe-8S-X]. The latter has a cage like structure, and it has recently been found that there is an X atom inside, which is believed to be an N in a resting (intermediate) state, i.e. as a µ6-nitride.
NiFe Hydrogenases
Catalyses H2 oxidation in some bacteria, which use H2 as a fuel. It is very active, and may be able to replace Pt as a catalyst for hydrogenation. This is an example of an Ni,Fe/RS cluster.
Catalysis
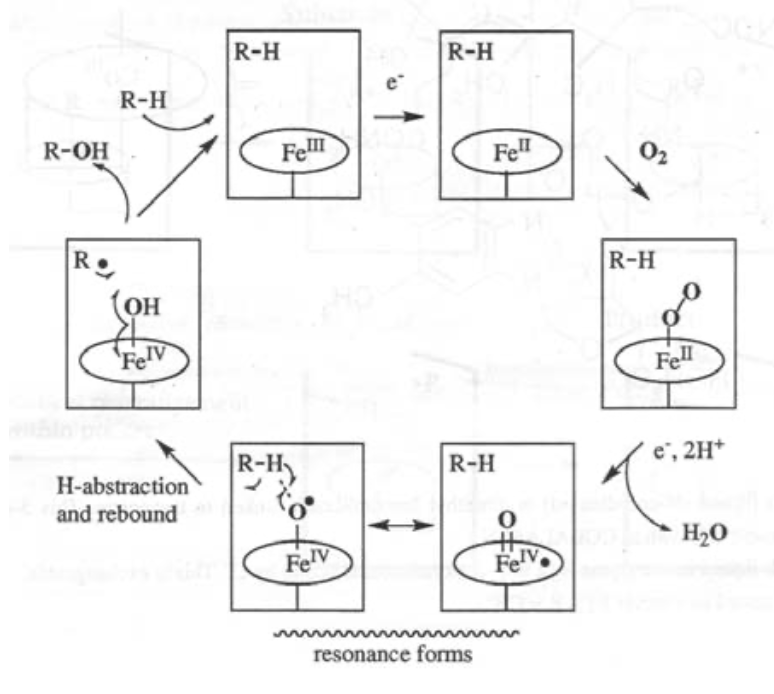
Fe(IV)=O is particularly relevant here, as it is strongly oxidising. It can be used as part of the O2 --> 2H2O process, and also in steroid synthesis and destroying toxins.
The classic example of Iron catalysis is Cytochrome P450:
Fe-Transport Proteins
Transferrins
These are glycoproteins, MW ~ 80000. There are 2 separate and approximately equivalent Fe binding sites.
They complex Fe(III) (which is hazardous when free), with simultaneous binding of HCO3- or CO32-, and release of H+. KA for the process is about 1023, but this is highly pH dependent, which controls the uptake.
A conformational change occurs on binding, locking the Fe(III) in due to the loss of internal mobility.
To release it again, the pH must drop, and something must be present to pick up the Fe(III) as it leaves. This is a chelator, such as siderophores, or a transferrin receptor on the cell surface (which then enters by exocytosis).
Ferritin -
This stores non-heme iron in animals. It can carry 20% Fe by mass! It is a very large (MW 460000 - 550000) spherical protein, and is present in all types of organism. In mammals, it is typically found in the spleen and blood.
It is made up of 2 components - a “mineral” core (up to 4500 Fe atoms) and a protein shell. The 24 subunits that make it up link together like a soccer ball into a hollow sphere. Each subunit has 4 long α-helices and 1 short one, and then an anti-parallel β-sheet.
The mineral core comprises hydrated Fe(III) oxide + phosphate. It resembles 5Fe2O3.9H2O, with a HCP array of O and OH, with Fe3+ in the octahedral and tetrahedral holes. There is significant antiferromagnetic coupling. We cannot currently see where the phosphates are in relation to this (transparent to X-rays due to random ordering).
The iron enters and leaves through a controlled process:
2Fe(II) + O2 + 2H+ <=> 2Fe(III) + H2O2
This reaction occurs via an inner-sphere electron transfer mechanism at ferroxidase centres. Oxidation allows the iron to enter, while reduction allows it to leave.
Copper
Cu Centres
Copper has useful redox properties, although for this reason it is extremely toxic when not bound to ligands. It usually binds to two histidine ligands, and then a cysteine if taking part in electron transfer. The maximum coordination is usually 5. There are no macrocyclic ligands known for it. Generally it is extracellular (unlike Fe) - presumably due to its toxicity. Also it appeared later in evolution due to solubilities relative to Fe.
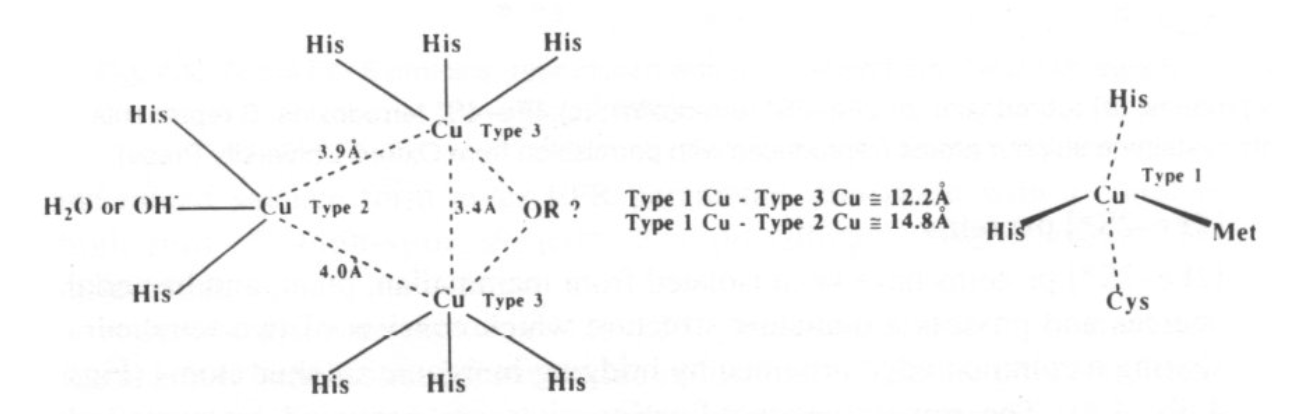
Electron-Transfer in “Blue Cu Centres” -
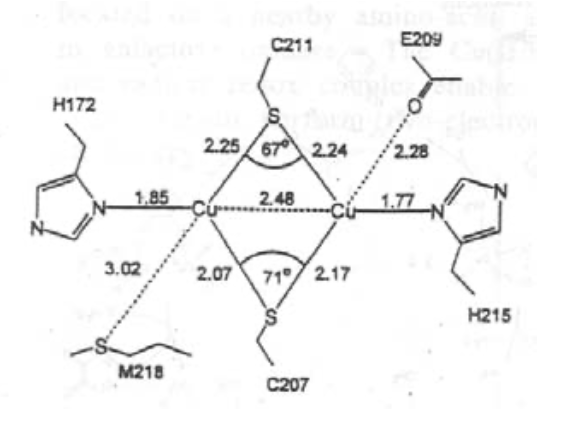
This is very fast, as Cu(II)/Cu(I) are in the same geometries due to the entatic state.
Trigonal pyramidal geometry is usually seen, with 3 close equatorial ligands (2xHis + Cys) and then 2 axial (Gly and Met usually).
Binuclear Cu2 centres have been discovered in cytochrome c oxidase and N2O reductase. These were determined by spectroscopic and X- ray data.
It is believed to act as a relay centre (for e-transfer).
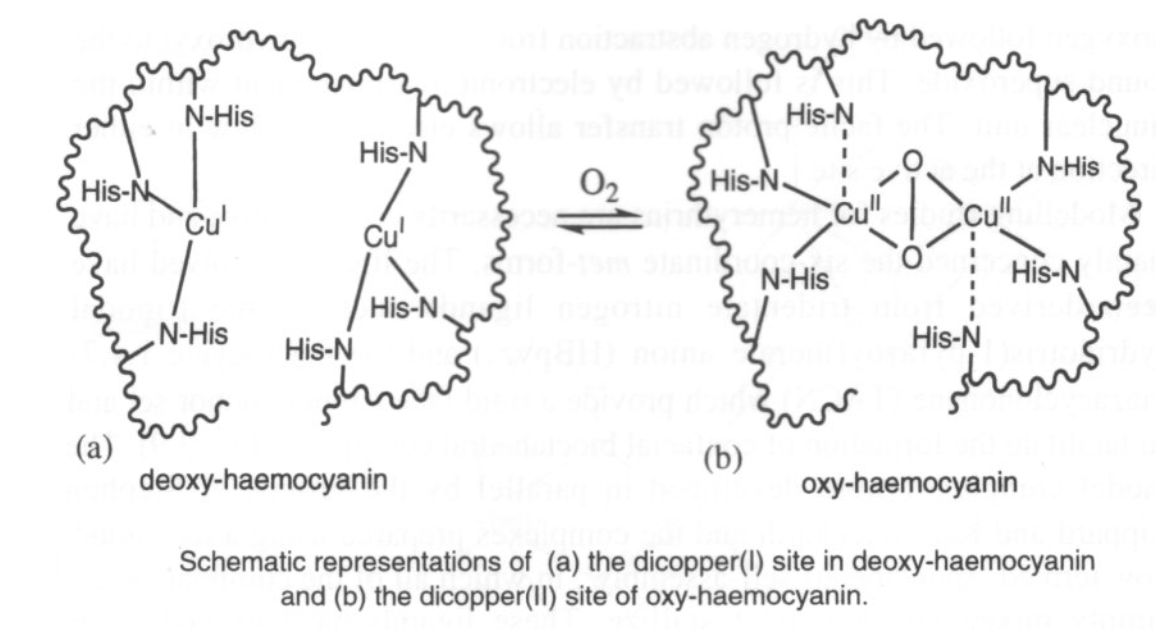
Reversible O2 Carriers
Hemocyanin (in arthropods and molluscs) has a binuclear Cu site too. It binds O2 reversibly (as in haemoglobin) although it is much harder to remove, and therefore a higher energy process. It is generally used for organisms where O2 is scarce (aquatic environments, for example) where as much O2 has to be taken in as possible.
O2 only binds to the Cu(I) form. However, charge transfer onto O2 occurs, so Cu(II) forms. This also gives rise to the blue colour.
Ascorbate Oxidase
This turns poly-ols into poly-als on the skins of fruit, via a poly-ol radical. This product then polymerises to protect the fruit from O2.
The active site is a trinuclear Cu centre, with a blue Cu centre near the surface that supplies the former with electrons.
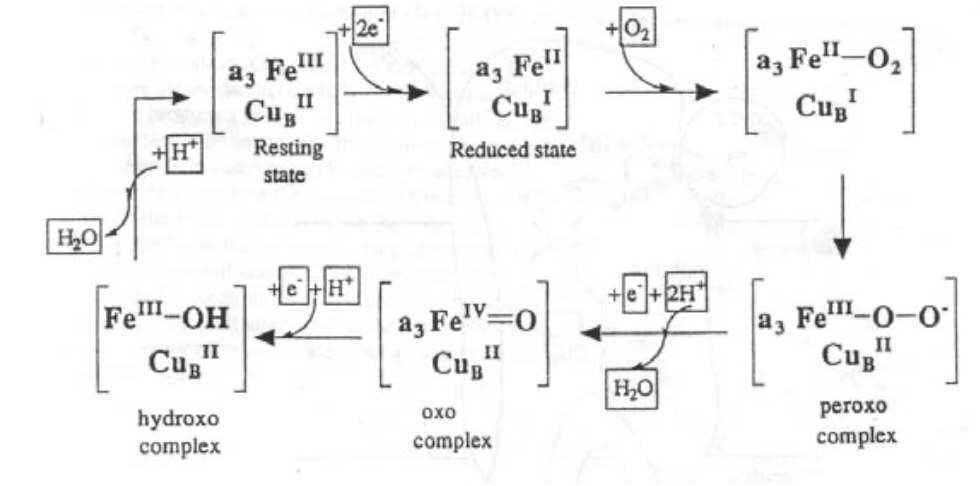
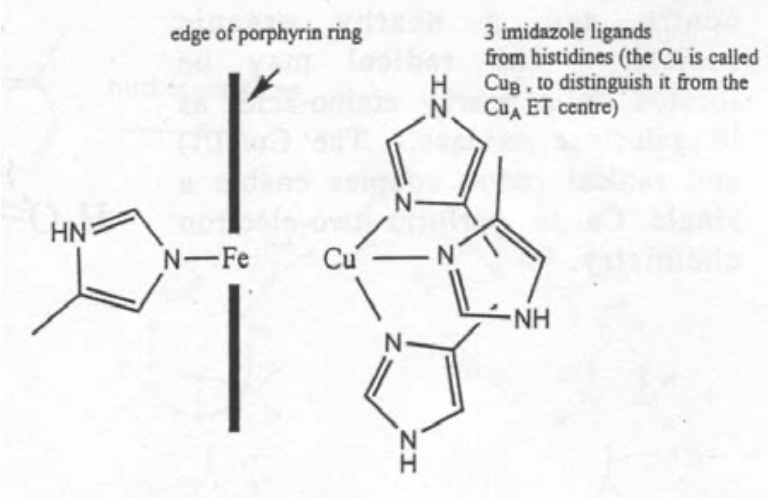
Cytochrome c Oxidase
This is found in the inner mitochondrial membrane, and is vital for aerobic organisms. It catalyses the reaction O2 --> 2H2O using 4 electrons, which come from oxidation of “CHO” and are delivered by Cytochrome c.

The active site is like a cross between haemoglobin and half haemocyanin.
It works with no harmful O-intermediates, and the energy released is used efficiently to pump H+, which sets up a gradient that drives ATP formation from ADP).
Cobalt
Coenzyme B12
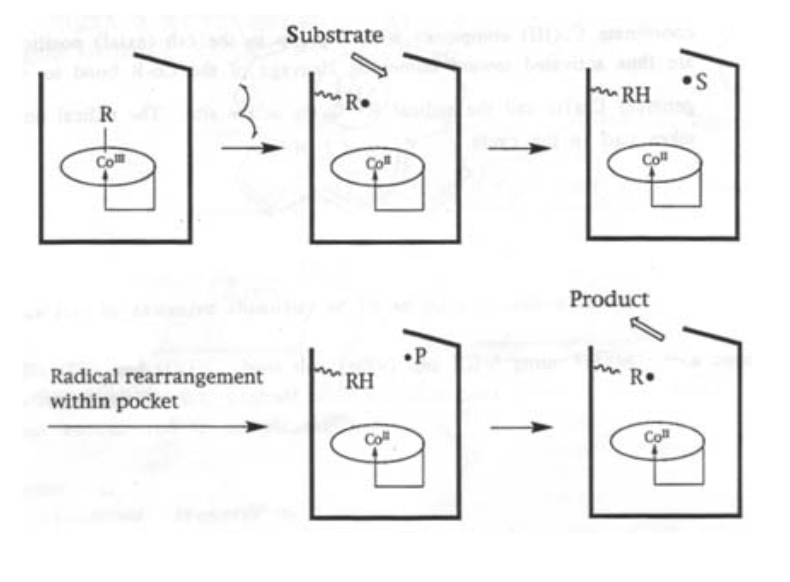
This is a cobalt-containing cofactor, and catalyses specific radical-based organic reactions (e.g. isomerisations) when bound to the appropriate enzyme.
It is a macrocyclic complex with a Co-alkyl bond. It is also the only known biologically active role for Cobalt, and one of very few organometallic compounds in nature.
It can only be synthesised by micro-organisms, but humans require trace amounts.
The macrocycle is a corrin ring - this is similar to the porphyrin, but only has 15 sides instead of 16. It also then coordinates an N-ligand (dimethyl benzimidazole) and 5’-deoxyadenosine by C (this is replaced by -CN for ingestion).
There are 3 oxidation states possible for the cobalt, all of which are low spin. CoII has an unpaired electron in a dz2 orbital (shown by EPR), which is the key to its catalytic properties.
Zinc
Acid-Base Catalysis
Zn(II) is good because it binds strongly to proteins, while allowing rapid ligand exchange (e.g. for H2O or substrate). It also has good electron affinity and flexible geometry, with no redox chemistry troubles.
This helps biology find away round the need for strong H+/-OH to catalyse reactions (which would lead to hydrolysis of most biological compounds!).
Zn-carbonyl mechanism:
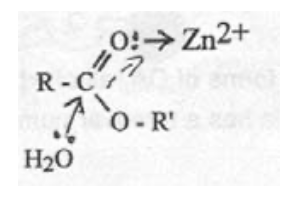
Zn-hydroxide mechanism:
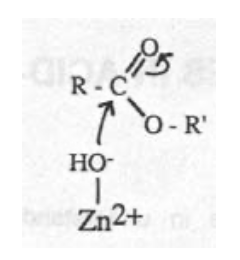
Carbonic Anhydrase (CA)
CO2 + 2H2O <=> HCO3- + H3O+
Turnover of CO2 in biological systems is high, but solubility (this equilibrium) is very slow, and needs to be catalysed.
CA II has a MW ~ 30000, and is found in red blood cells. It carries this out at 106s-1, and is one of the most active of all enzymes.
The geometry around Zinc is a distorted tetrahedron, with 3 His ligands and OH2. It operates by the Zn-hydroxide mechanism, and delivers OH into CO2.
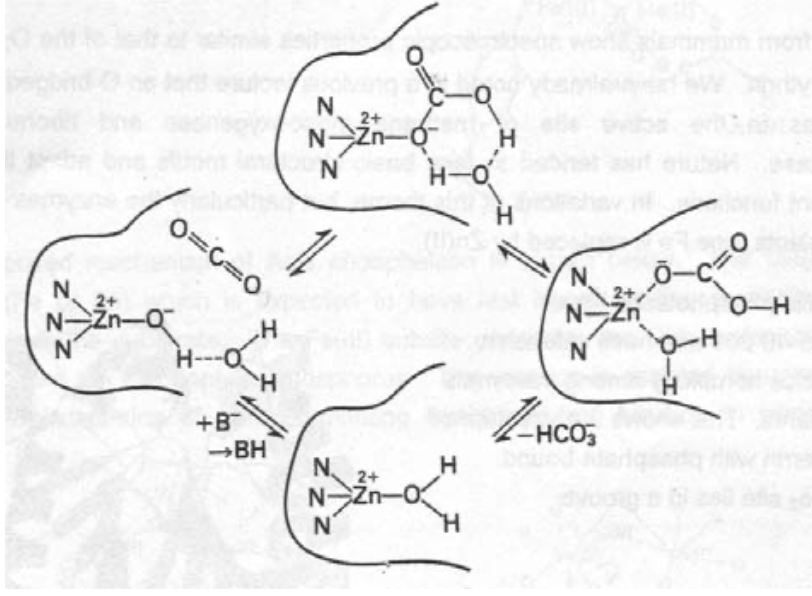
Carboxypeptidase (CPD)
Carries out peptide hydrolysis. There are 2 types of Zn (A & B). B acts on basic residues, while A acts on C-terminal amino acids, and is thus easier to study.
CPD A has a MW of 34600. The Zn is found in one side of the groove where the substrate binds, and is coordinated by 2 x His, water, and a Glu residue (bidentate). It can however go up to 6 coordinate, e.g. when the inhibitor glycyl-L-tyrosine binds (with NH2 and carbonyl-O).
Alcohol Dehydrogenase (ADH) -
MW ~ 80000. Oxidises primary and secondary alcohols to aldehydes / ketones. Uses NAD+ as e-/H+ acceptor, and so is a redox enzyme. The Zinc activates the substrate for reaction with the cofactor. A subunit has two Zn atoms - one catalyses while the other stabilises the structure. Catalytic Zn has 2xCys + His + H2O coordination, while the structural Zinc has 4xCys protein ligands bound to it.
Alkaline Phosphatase -
Hydrolyses phosphate esters (inc. ATP), and works best under mild alkali conditions.
R-OPO32- + H2O --> ROH + HOPO32-
It is a dimer, and each subunit has two Zn atoms in close proximity. The Phosphate then bridges the 2 Zn atoms. The other ligands are His and Asp.
Calcium
Calmodulin
Molecular Weight of ~ 17000. Has four Ca2+ binding sites in 2 pairs, at each end of a long molecule. Folds are called “EF Hands”. Kd for binding: 10-5-106 mol dm-3.
The binding alters the conformation, therefore it is recognised by enzymes (protein kinases). These are then activated to catalyse the phosphorylation of proteins.
TUTORIAL QUESTIONS - CYTOCHROME C, FE VS. CU, & COORDINATION CHEMISTRY
Discuss the validity of the statement “Cytochrome c is perfectly designed for rapid electron transfer”.
Cytochromes are heme proteins which act as 1 electron carriers. They are found in all forms of aerobic life, as they are essential for the transformation of O2 into water, which drives the formation of ATP (the energy source of aerobic organisms) by a proton gradient across a membrane.
They can be tailored to suit the environments they are placed in by the organism, and broadly speaking there are 4 groups, labelled a-d. Cytochrome c features covalent links between the heme group and the protein, and is one of the best characterised and most studied of all enzymes due to the fact that it is readily soluble from the membrane.

Cytochrome c occupies a position in chain of different cytochromes, and the whole process catalyses the formation of ATP from ADP. The chain is something like this:
NAD+ --> Flavoprotein --> Cyt b --> Cyt c1 --> Cyt c --> Cyt c oxidase --> O2
It also has two strong field ligands (Histidine and Methionine), so the iron is in its low spin configuration. This is advantageous for electron transfer as t2g6 <--> t2g5 (outer sphere) exchange will be far easier than any involving antibonding eg orbitals (the latter would require more distortion of the coordination sphere according to Marcus Theory, which expends energy and therefore slows the reaction).
Analysis of the crystal structure of cytochrome c containing both Fe(III) and Fe(II) has been accomplished. The structures are almost identical, which is further indication that the process is very fast, as a lack of change in conformation is ideal according to the Franck-Condon Principle (that is that electron transfer must occur between complexes of the same energy). This means that there is no need to adjust bond lengths between the two forms so that their energies are equivalent, i.e. that the free energy of reorganisation is very small. This results in very fast reactions according to Marcus Theory.
Outer Sphere - energy terms:
Form Encounter Complex Æ Reorganisation Energy + ∆G for the process.
The initial formation of the encounter complex is essentially protein chemistry, and irrelevant of the metal ion. The reorganisation energy is the key, and is governed by Marcus Theory (self-exchange rates and equilibrium constant (∆G)). It is composed of ligand and solvent reorganisations.
The ligand reorganisation depends on both covalent (CFSE) and ionic (charge) contributions, while the solvent reorganisation depends on charge and ligand size (latter affects the size of the hydration sphere).
Cytochrome is almost perfectly designed to maximise these effects. It has large, fixed ligands (heme) which greatly reduce the solvent reorganisation energy term. The ligands are also uncharged for this also. The t2g6 <--> t2g5 exchange also is a non-bonding exchange, therefore less covalent effects in operation. Thus, the only ligand reorganisation is monopolar/dipolar axial ligands.
In terms of catalytic activity then, we can expect the rate to be very fast. This is enhanced by the π-system (porphyrin + phenylalanine close by). Recent evidence has suggested that this is not actually completely necessary though. A side-effect of this is that the electron transfer occurs over some distance, and it would be more efficient to have the transfer occurring between species as close as possible to each other (e.g. iron clusters). This then becomes a balance between reorganisation energy of ligands and close site transfer.
∆G is also important to consider. We want to minimise this, as we need to go down an energy gradient at the correct rate to pump protons most efficiently. The key point is not speed - thermodynamic constraint within which we want to go as fast as possible.
The structure also indicates that the iron is fully coordinated. This shows that the protein’s sole role is for electron transfer - no other reactions occur at its active site. This also renders it immune to other complexing ligands that may be toxic, e.g. cyanide, which deactivates cytochrome a. Another advantageous property is that the reaction is very clean. There are no other side-reactions and no harmful by-products or hazardous O-intermediates.
The mechanism by which the enzyme moves between the reductase (Cyt c1) and the oxidase (Cyt c oxidase) is not well understood. It is believed that when docking with the reductase, an electron is donated which is passed to the heme ring by π-electron cloud overlap, which gives rise to an ion pair. The fact that this is in a hydrophobic environment means that this is a very high energy intermediate, and this drives a conformational change that allows the electron to be shuttled between parallel residues, and compacts the protein and move to the oxidase.
Hence I would conclude that cytochrome c is perfectly designed for the role it carries out. It is fast and efficient and able to transfer large amounts of energy in a controlled manner over long distances. It’s only fault therefore would be that it requires many other components and enzymes in the electron transfer chain to be functioning correctly as well for it to actually be useful.
Compare and Contrast the roles of Fe and Cu in (i) Oxygen Binding Proteins, and (ii) Oxidases
Iron has two oxidation states in solution, Fe(II) and Fe(III). Ferrous ions are less common as it is easily oxidised to ferric. Most of the iron in humans is found at the centre of haemoglobin molecules, although some is stored in the liver and the rest used in redox reactions and protein formation in the plasma. The type of ligands attached greatly affects the properties (as would be expected). Typically, it is found as divalent (haemoglobin), trivalent (catalases and oxidases) or oxidised and reduced between both states (cytochromes).
Copper is also found in two different oxidation states, cuprous (I) and cupric (II). The former is easily oxidised to the latter in the same way as for iron. Cu(I) usually has coordination numbers equal to or less than 4, while Cu(II) reaches 6 (albeit Jahn-Teller distorted) and is rarely below 4. Next to iron, cupric is the best catalyst in oxidation-reduction processes. It is found in enzymes such as phenolase or hemocyanin, and these carry oxygen in the same way as haemoglobin. It is also required in the production of haemoglobin.
Fe tends to be found inside cells, while many Cu-containing enzymes are extracellular. Cu appeared in evolution far later than Fe, probably due to its greater availability in an aerobic environment (Cu2+ more soluble than Cu+, but Fe3+ are less soluble than Fe2+).
Fe is usually found inside a porphyrin-type macrocycle below.
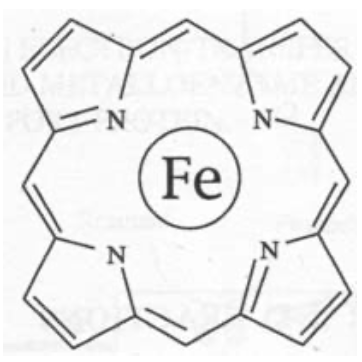
Cu is rarely found in macrocyclic ligands, and tends to favour coordination by two or more Histidine ligands, and often a cysteine in electron transfer reactions. This reflects its “softer” chemical nature (i.e. bonding shows a higher degree of covalency and overlap).
Reversible Ligand Binding is achieved by the O2 molecule binding to the Fe end-on, in a bent configuration. The diamagnetism of the complex to Fe(II) indicates that there is a rearrangement of spins. This has been suggested as singlet O2 bound to low-spin Fe(II), with strong preference over Fe(III). The complex becomes bent as the occupied π*-orbital on O2> donates and the empty π*-orbital accepts. It should be noted though that simple Fe-porphyrin compounds do not exhibit reversible O2 binding, as steric hindrance is required to prevent oxidation.
For copper-based reversible O2 carriers such as haemocyanin, a binuclear Cu site is often seen.
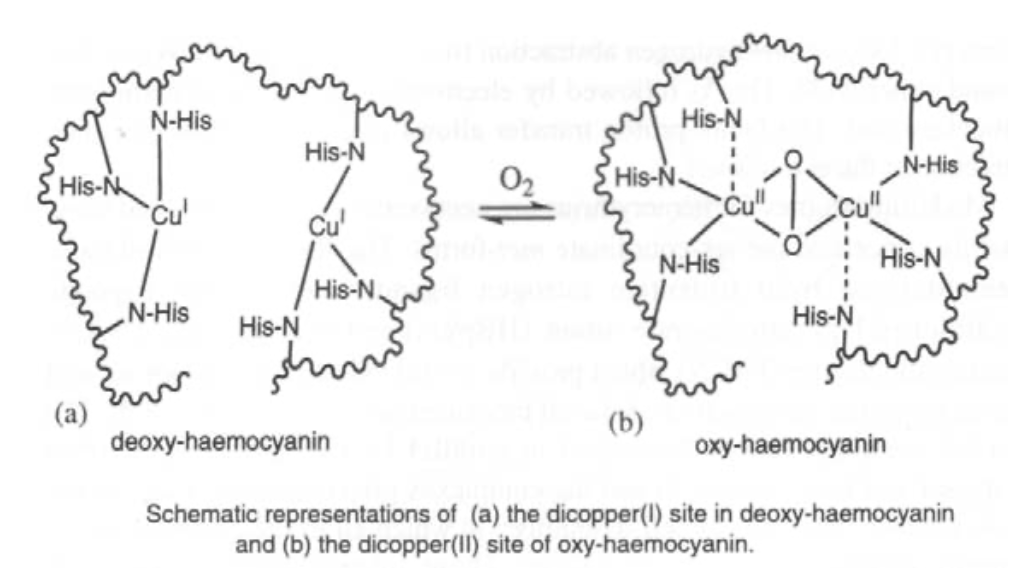
O2 is bound reversibly as in haemoglobin. Binding will only occur to the Cu(I) form, but there is extensive charge transfer onto O2 so that the two Cu atoms are oxidised to Cu(II). This gives it a blue appearance.
The Fe-O2 bond is weaker than the Cu-O2 bond. This is a good thing in an O2 rich environment to maximise unloading potential (i.e. need to be able to pick up and release it as efficiently as possible). Cu is generally found when less O2 is available, e.g. in water organisms. This is because they need to pick up O2 whenever they can. Hence they develop a mechanism for removing it from the Cu binding site, which occurs at a higher ∆G. This is wasteful of energy, unless it is necessary for their survival (hence it is not used in humans). Also, Cu is poisonous if not handled carefully by the process.
Electron Transfer reactions are similar to those seen for ordinary complexes, and the rates are governed by Marcus Theory. Thus, importance factors are the driving force, reorganisation energy and electronic coupling between donor and acceptor sites.
Electron Transfer chemistry requires the full range of electrode potentials in order to function, e.g. the electron transport chain starts at negative potentials and works its way up to positive potentials. This optimises Eo for the process. Hence, both Fe and Cu find roles in it, as they have potentials that are optimal for different portions of the chain. Eo(Fe2+/Fe3+) = +0.77V, while Eo(Cu+/Cu2+) = +1.1V. Hence, Cu(I) can perform O2 chemistry, and finds many of its uses in oxidases (see below). The only way for Fe to have enough potential to do this would be if it were the Fe3+/Fe4+ couple. This is also used sometimes, e.g. cytochrome P450 which uses the iron(IV) oxo complex. However, Iron(II) is used for more negative Eo processes in the Electron Transport Chain (i.e. near the start).
For iron, the porphyrin macrocycle serves as an electron antenna:
Particular examples of this would be the cytochromes (cytochrome c discussed earlier).
However, there is also a range of iron-sulphur cluster proteins, which are found in all living organisms, where they are involved in a wide range of electron transfer processes. The iron is bound by sulphur atoms, either from cysteinyl residues or by inorganic sulphide., and there are a considerable range of structural types. Examples of some of the geometries possible are shown on the left.
Copper electron transfer proteins usually involve “blue” Cu centres. These can also transfer electrons at very high rates. The protein enforces the same geometry on the Cu irrespective of whether it is Cu(II) or Cu(I), so the reorganisation energy is very small.
Oxidases typically show a range of Cu centres. Iron’s role is usually involved in electron transfer as discussed above.
The Cu centres can come in many forms, e.g. ascorbate oxidase has a trinuclear centre, which is supplied with electrons by long-range transfer from a blue Cu centre close to the enzyme’s surface.
Cytochrome c oxidase is however the most famous oxidase involving copper. It is found in the inner mitochondrial membrane and catalyses 4 electron reduction of O2 to two H2O. It is essential to all higher forms of life as it enables the use of O2 as an oxidant.

The site of O2 reduction has a Cu atom close to an Fe porphyrin. It basically resembles a cross between the active site of haemoglobin and one half of the active site of haemocyanin.
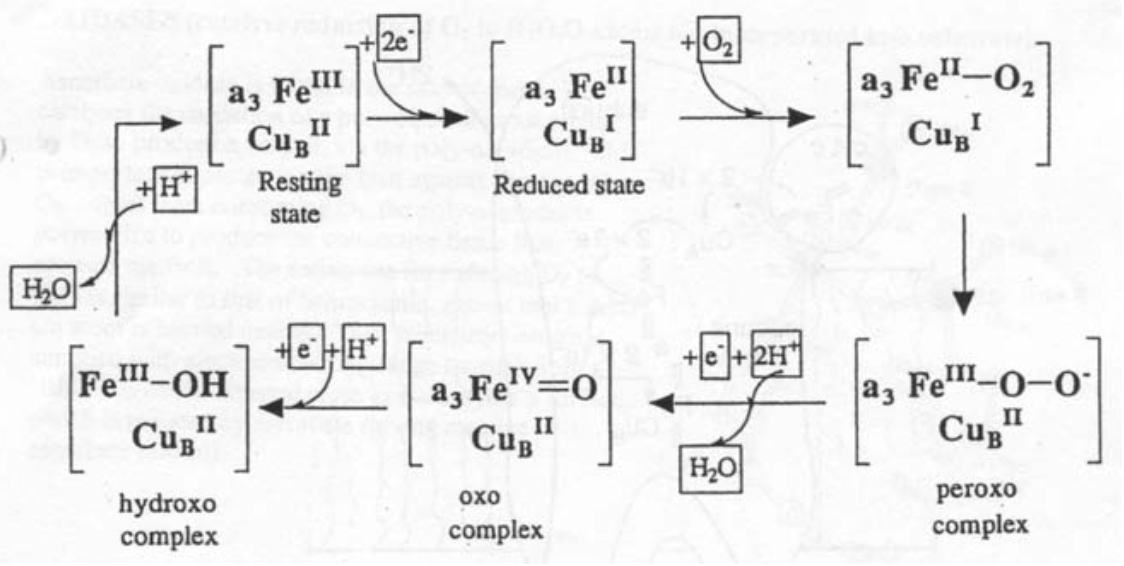
Thus, Fe and Cu are involved in the redox cycle:
In conclusion, both these elements show electron-transfer like properties on account of the sensitive equilibrium between the Fe(II)/Fe(III) and the Cu(I)/Cu(II) states respectively. They are also both capable of binding to O2, and thus provide a mechanism for O2 transport around organisms. Biologically the latter tends to be dominated by iron, probably because “it got there first” evolutionarily speaking, as a result of its solubility properties.
The coordination environments A to E sketched below are found for metal ions in certain proteins, and the two sites in E contain the same metal ion. Suggest which of the ions Mg2+, Ca2+, Fe3+ and Zn2+ is most likely to be associated with each environment, giving reasons for your choice. Note that one of the metal ions is found in more than one of these environments.
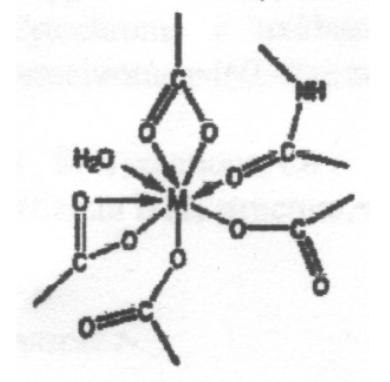
This is expected to be Ca2+, due to the high coordination number - Ca2+ is a large ion compared to the other metals (no d-block contraction) and so it can coordinate more ligands without excessive steric hindrance. Also, all the ligands bind through O, which suggests it is a hard ion (Ca2+ fits this assessment).
Group 1 & 2 metals --> maximum coordination limited by sterics as the only factors. Ionic Model.
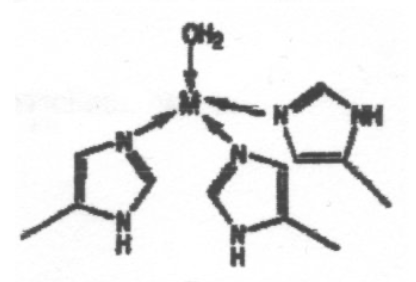
This is certainly Zn2+, and this particular arrangement is seen in Carbonic Anhydrase.
The distorted tetrahedron reflects Zn2+ small size (it sees the full effect of d-orbital contraction) so it can only coordinate 4 ligands. It also binds to N preferably to O (slightly softer).
4s/4p involvement (like p-block) --> Td.
Can go 5-coordinate (no CFSE issue) and 6 coordinate only with very small ligands.
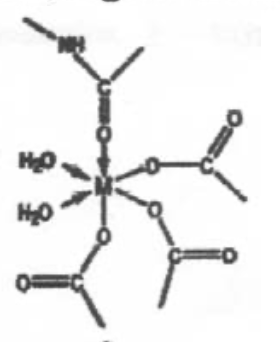
This is a perfectly regular 6-coordinate metal centre, with O-donor ligands. This is probably Mg2+, which is small, hard and polarising, and unlikely to distort (no d-electrons, so not redox catalyst).
Same argument as for A (Calcium). Ionic Model.
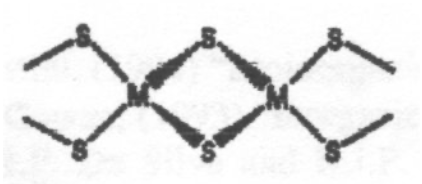
This is a typical arrangement for an Fe-S cluster. This particular one is 2Fe-2S. Tetrahedral coordination around Fe(III) or Fe(II). Electron Transfer catalyst. Increased covalency for S.
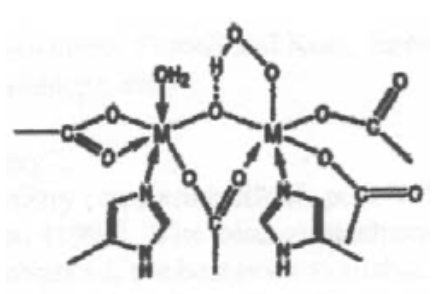
This is haemoerythrin, which indicates it is Iron.
N/O ligands favour 6 coordination (smaller).
5/7 coordination unfavourable - can’t maximise CFSE or covalent overlap.
Transition Metals are often governed by directional bonding and CFSE arguments, in addition to the electrostatic/steric arguments given by the Ionic Model.
Can control whether Mg2+ or Ca2+ binds in the same site. Obviously can make site too small for Ca2+ to fit in, but to stop Mg2+ entering Ca2+ site is more complex.
First we consider that the electrostatic (binding) potential varies as 1/r. Hence, with Mg2+ inside a cavity (with walls essentially oppositely charged), shifting the Mg2+ from the centre means that it will bind more strongly, due to the 1/r dependence (think about the graph - 2 points on either side of the mid-point have a larger total than 2 x mid-point). However, Mg2+ is much more greatly hydrated than Ca2+ (i.e. ∆Hhyd(Mg2+) >> ∆Hhyd(Ca2+), even though ∆Hbind for Mg2+ > Ca2+). Therefore make sure that binding energy doesn’t compensate for ∆Hhyd(Mg2+).
I am happy for them to be reproduced, but please include credit to this website if you're putting them somewhere public please!