Aromatic and Heterocyclic Chemistry
Covers aromatic reaction types, then moves onto the Synthesis and Reaction Types of each class of Heterocyclic Compound. Mostly from the two primers on these topics.
Aromatic & Heterocyclic Chemistry
Aromatic Chemistry
Aromaticity
This confers an energetic stability over the equivalent double bond system. This can be explained from an MO point of view. The Huckel Rule states that planar monocyclic conjugated hydrocarbons are aromatic when the ring contains (4n+2)π electrons.
Huckel MO Theory (HMO) is used for conjugated planar molecules, both cyclic and acyclic. It is based on the approximation that the σ framework does not interact with the π-orbitals (orthogonal), and it can be used to calculate relative energies of MOs.
Aromaticity –
- Planar, fully conjugated, cyclic polyenes.
- Generally more stable than their acyclic analogues.
- As the number of π electrons increases, generally get more reactive.
- Bonds normally of nearly the same length.
- In a magnetic field, a ring current is set up (observable by NMR).
- Ability to undergo electrophilic substitutions.
Electrophilic Substitution
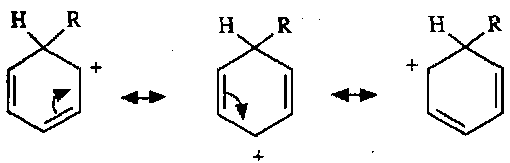
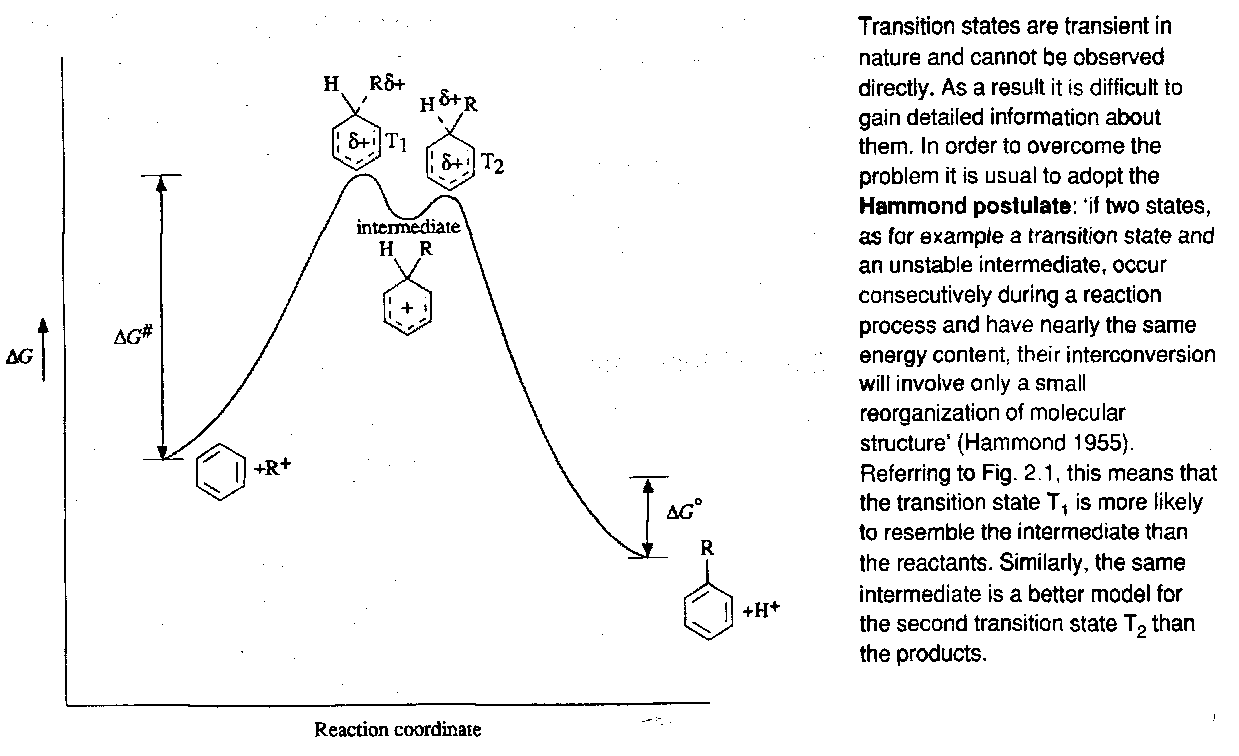
Evidence for this mechanism lies in the isolation of intermediates, and also kinetic isotope effects (Step 1 is usually rate determining).
Nitration – HNO3 + 2H2SO4 forms NO2+.
Sulphonation – H2SO4 forms SO3 at 80oC.
Halogenation – X2 + Lewis Acid.
Alkylation and Acylation – Friedel-Crafts Reaction. Alkyl/Acyl Chloride + AlCl3.
Note that alkylation typically runs to completion by substituting three times, whereas acylation shows less tendency to do this.
Formylation can be effected by adding HCl and CO with catalytic AlCl3. Other methods include:
Vilsmeier Reaction
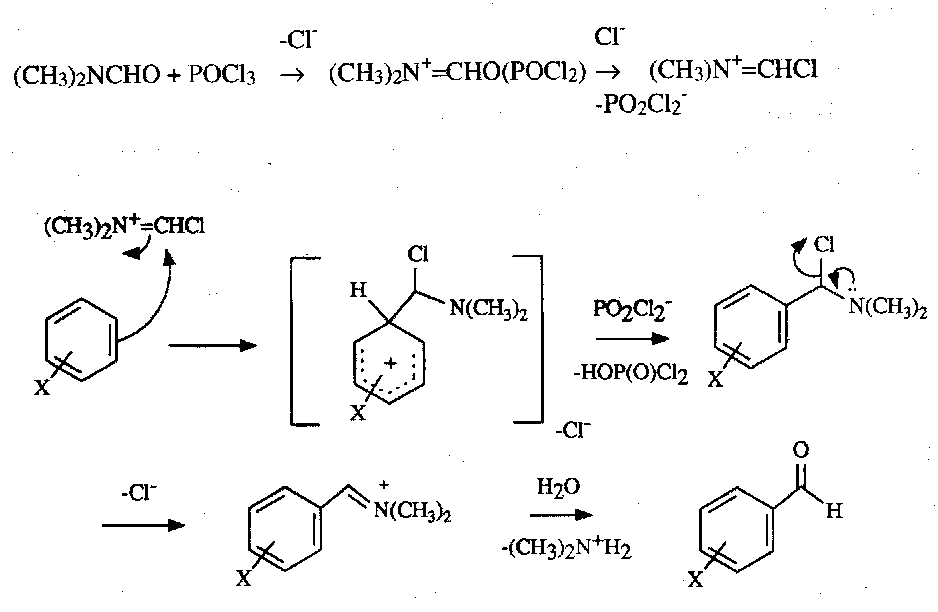
Hoesch Synthesis

Regiochemistry
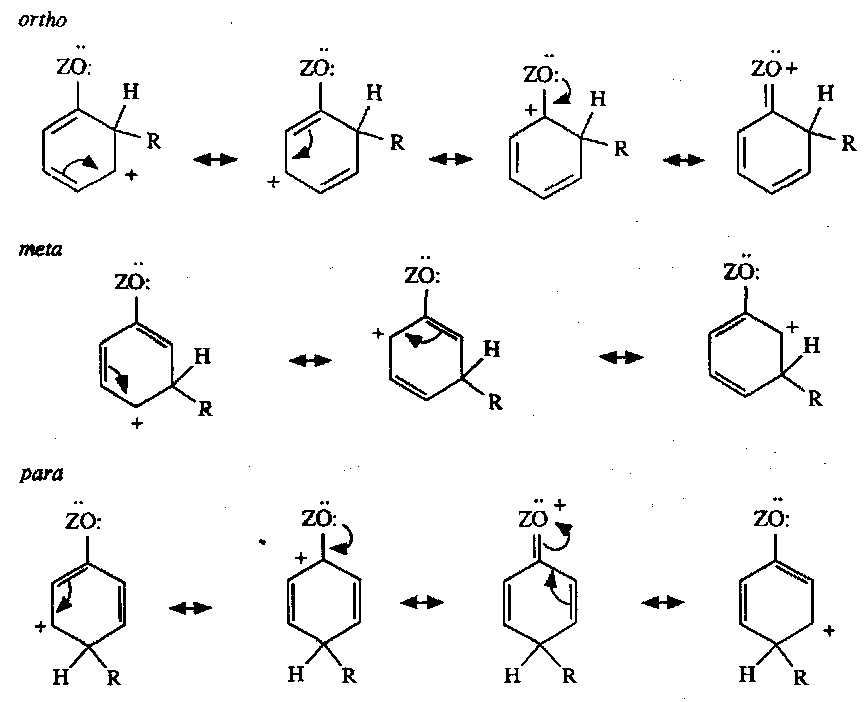
From a purely statistical point of view we might expect the ratio of ortho:meta:para products to be 2:2:1 based on the number of available sites. However, the nature of the substituent group has a major effect on the ratio of products.
Electron-withdrawal by a halide (Inductive Effect, -I) has a slight effect, but it diminishes rapidly with distance, and is typically outweighed by mesomeric (resonance) effects. The same is true for electron-donation (slight with alkyls).
Mesomeric effects can be electron donating or electron withdrawing as well.
We see that for electron withdrawing groups, this favours meta.

A positive charge is placed next to the ortho and para positions, so the internal energies for these intermediates will be higher (i.e. the reaction profile has higher energy transition states) than that of the meta, and they are formed more slowly.
In contrast, mesomerically donating groups share the positive charge at the ortho and para positions more effectively than at the meta, so their energy profiles are lower in energy than for the meta, and so favoured.
Ipso Substitution
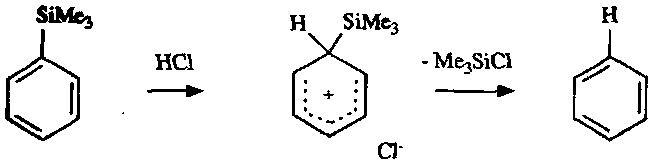
Summary of Substituent Effects
For EDG – generally ortho/para directing. All activated wrt benzene.
For EWG – meta directing. Deactivating wrt benzene.
Steric Factors
All things being equal, a third group is least likely to enter between two meta groups.
Ortho Rule: when an m-directing group is meta to an o/p-directing group, then electrophile goes ortho to the m-directing group rather than para.
Other Selectivity
Worth remembering Kinetic vs. Thermodynamic Control, especially with fused rings (disruption of both rings’ aromaticity vs. steric effects in the product).
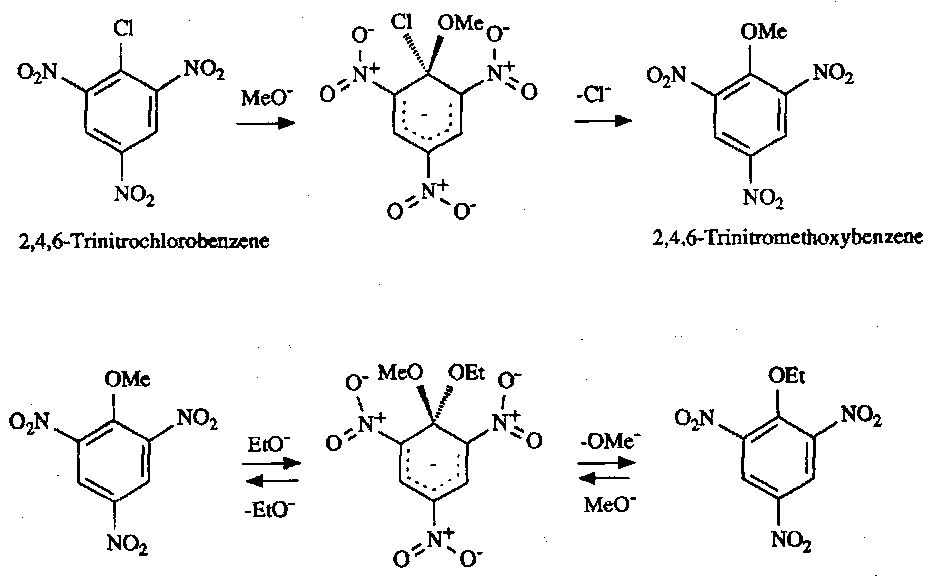
Nucleophilic Substitution
Requires strong electron withdrawal from the ring for initial attack. The intermediate is often called the Meisenheimer (σ) complex, and the Nucleophile attacking is rate determining.
Aryne Formation
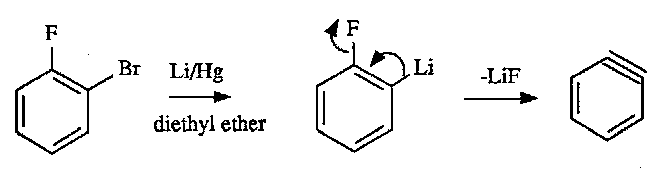
Can also deprotonate the ring with strong base such as –NH2 (imagine H in place of F in the above) giving rise to the same benzyne intermediate (highly reactive). In the case of –NH2 deprotonation the NH2 typically acts as a nucleophile attacking the benzyne intermediate (at either end of the triple bond). This is typically called cine substitution.
Unimolecular
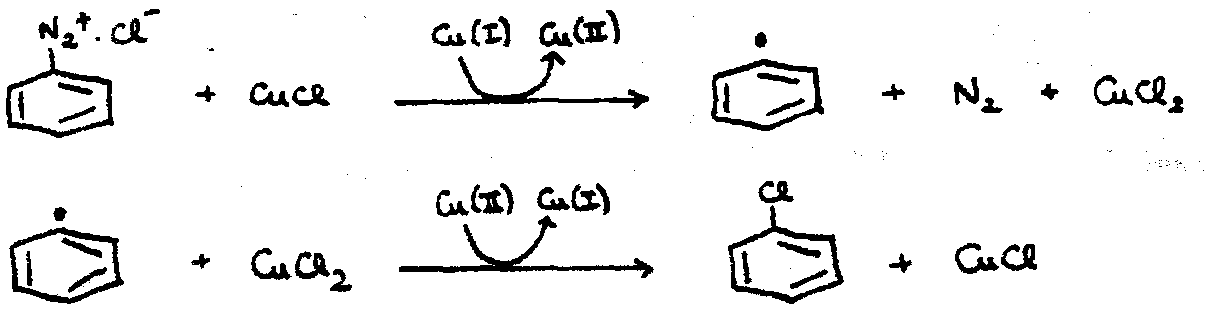
This occurs e.g. with diazonium salts, where the N2 falls off the benzene ring (rate determining), leaving C6H5+, which is then attacked by a nucleophile (fast).
Radical Reactions
Ph• formation
Via Electron Transfer
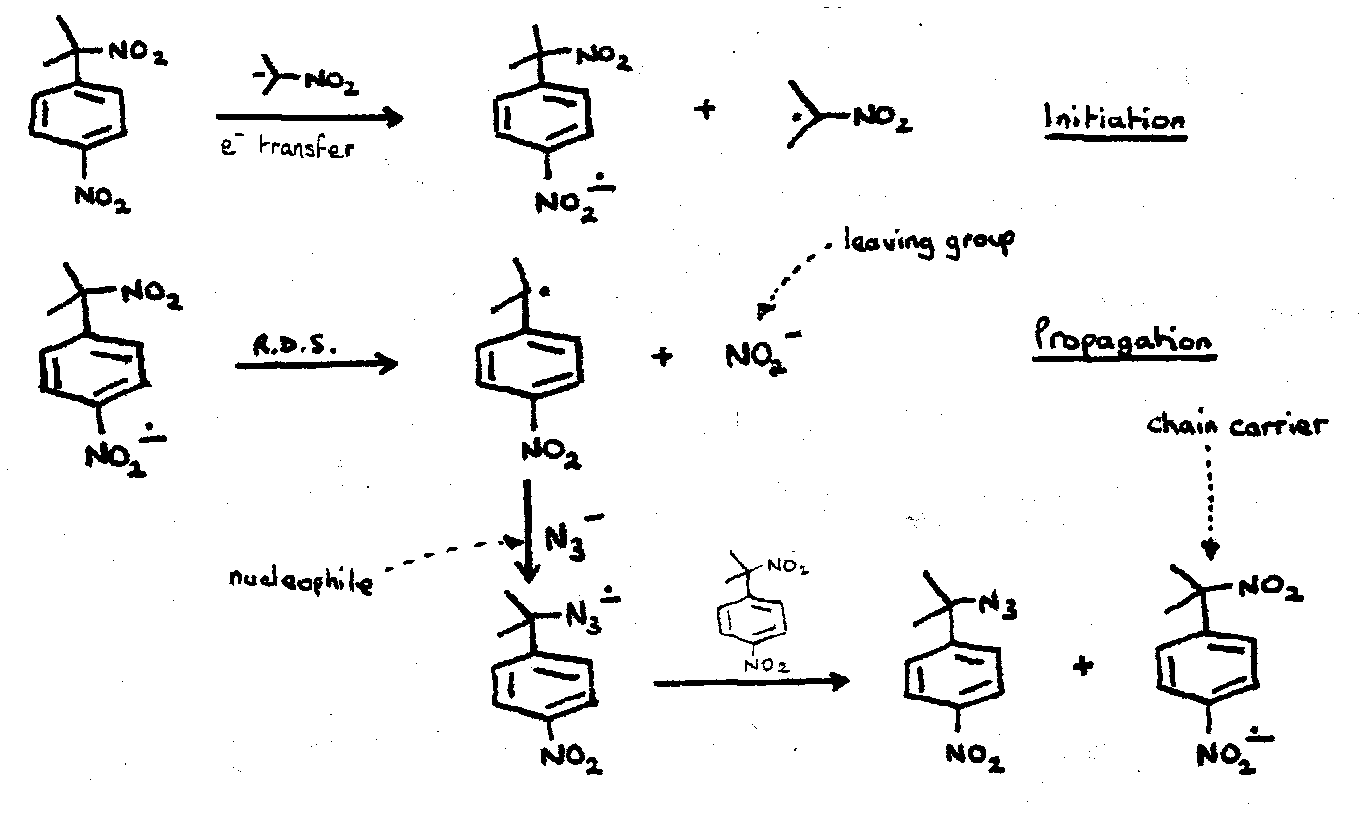
e.g. the SRN1 mechanism.
Initiation: e- transfer to the substrate.
Ar-X + e- → Ar-X-• → Ar• + X-
Propagation: process becomes a chain reaction if radical generated by leaving group expulsion reacts with nucleophile to give a radical anion capable of sustaining the chain.
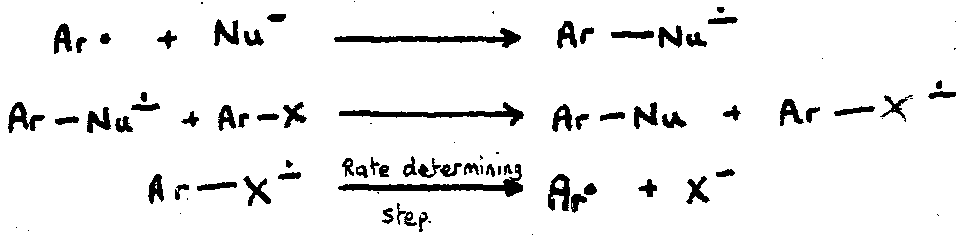
e.g.
Electrocyclic Rearrangement
e.g. the Claisen Rearrangement:
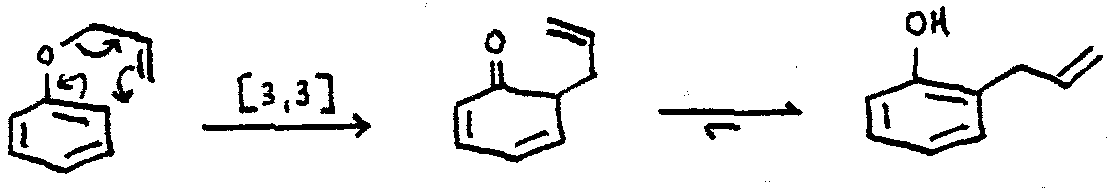
Note that there is also an electrocyclic step in the Fischer Indole Synthesis.
Compounds and Reactivity
Aryl Halides
These are most useful when converted into Grignard Reagents. They can then undergo a wide variety of transformations:
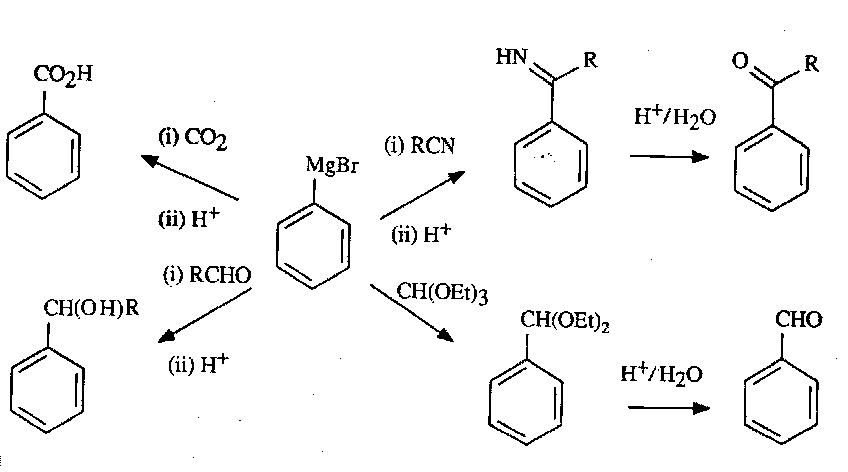
Reactions with Metals
Ortho-Lithiation
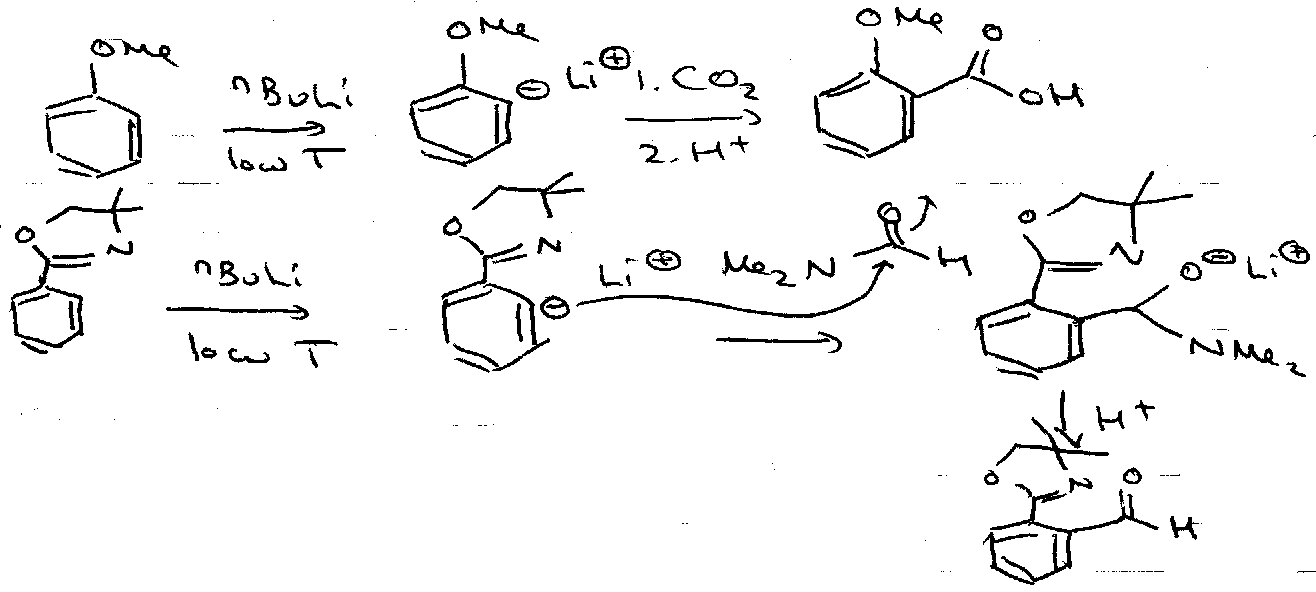
Features –
- Aryl sp2 carbanion out of plane of aromaticity – no resonance stabilisation.
- sp3 carbanion (Bu-) → sp2 carbanion (Ar-).
- Aryl- also stabilised by inductive (-I) effects.
- EWG often have a heteroatom in position to form a 5 or 6 membered intermediate – helps stabilise Ar-.
- Kinetic effects – Li+ counterion coordinates to substituent lone pair electrons – direct deprotonation and chelation control.
Palladium-Catalysed Cross-Couplings
Heck Reaction

See Named Reactions Cards for mechanism.
Stille Reaction
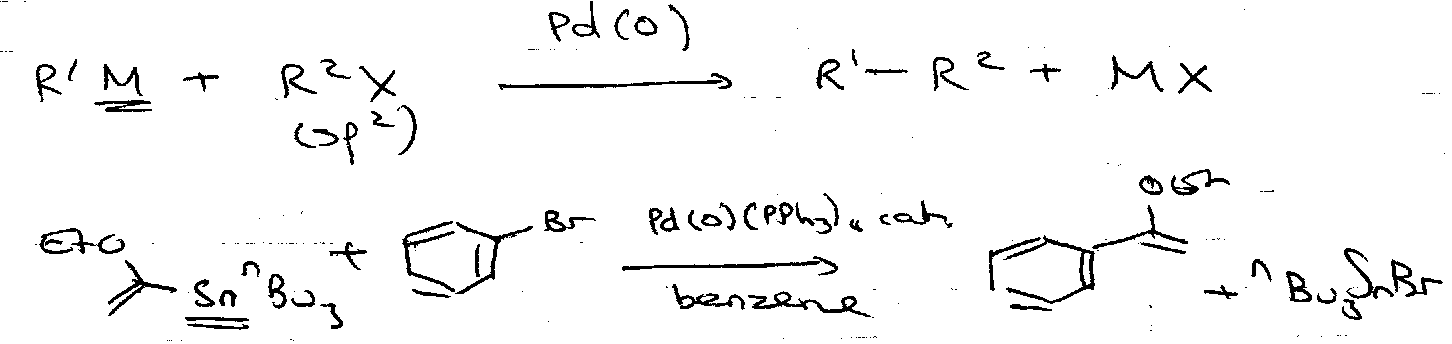
See Named Reactions Cards for mechanism.
Suzuki Reaction

See Named Reactions Cards for mechanism.
Chromium Complexes
General Properties – Cr complexes can achieve reactions that are impossible for free arenes, e.g. enhanced nucleophilic addition.
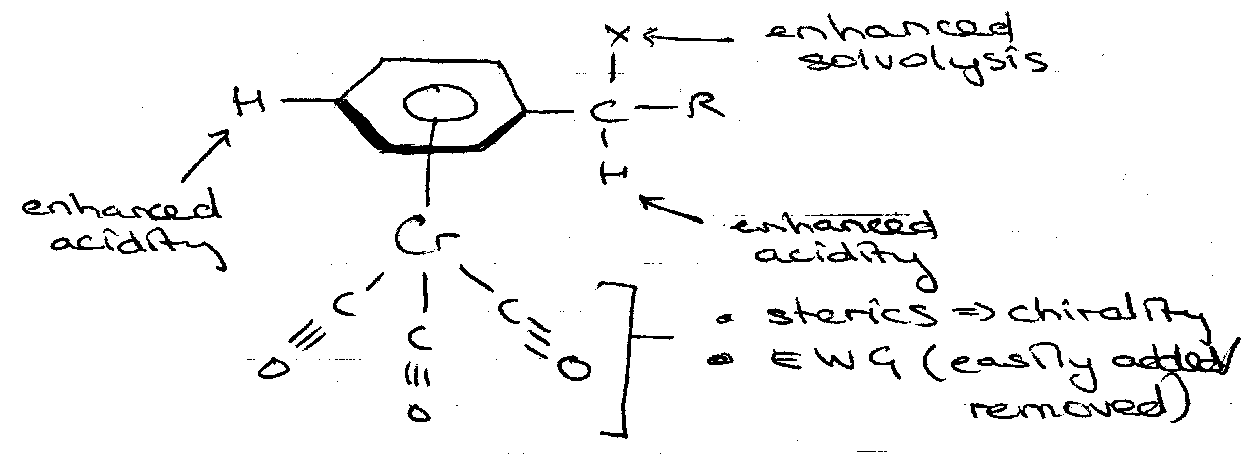
Nucleophilic Addition
- Unsubstituted
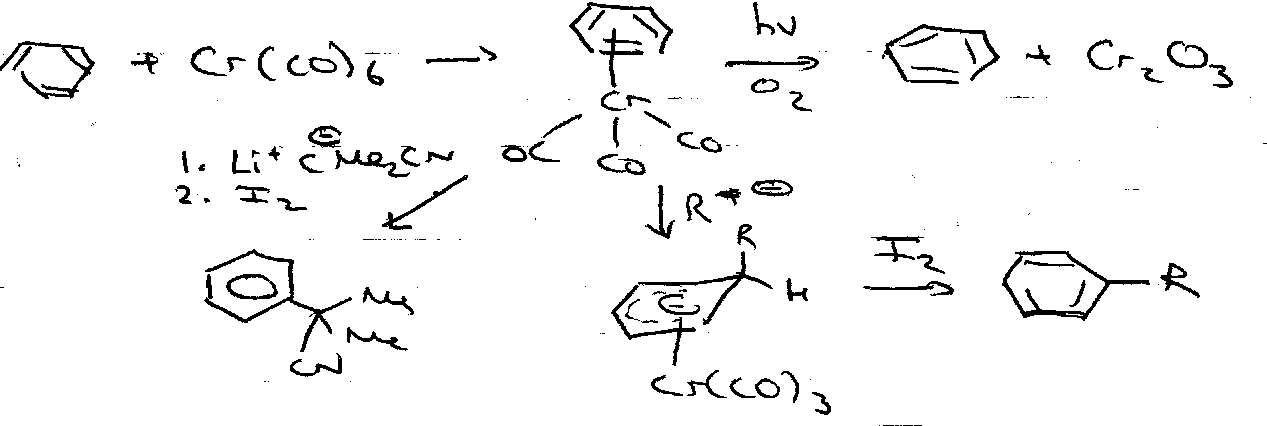
- Substituted
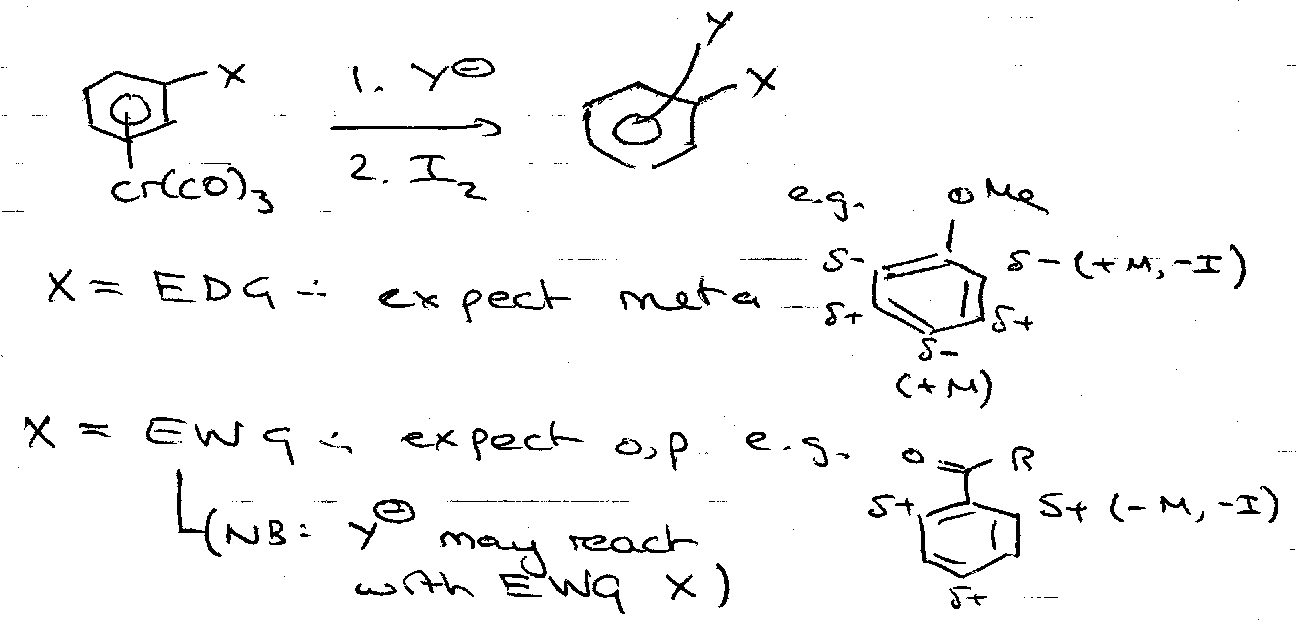
Nucleophilic Displacement of Halogens (addition/elimination)
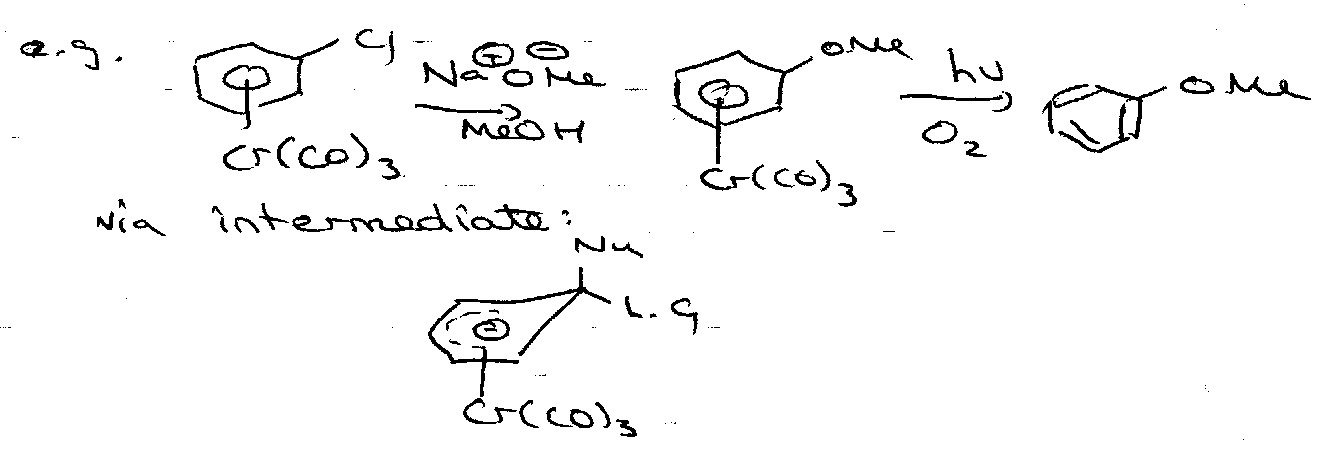
Heterocyclic Chemistry
Pyrroles, Thiophenes and Furans
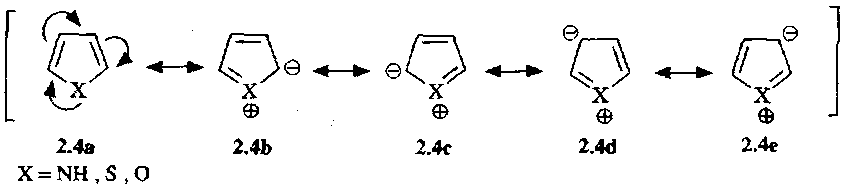
Synthesis
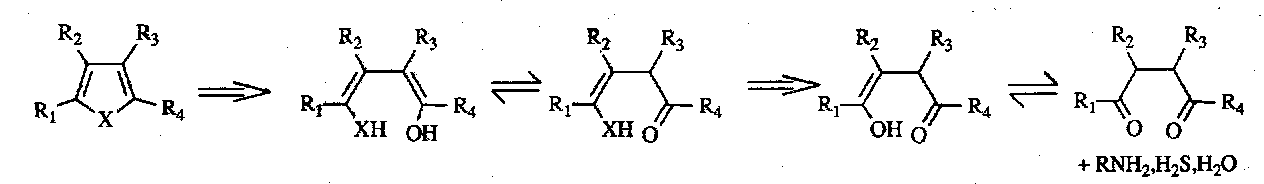
The forward process is known as the Paal-Knorr Synthesis. This is a very straightforward synthesis limited only by the accessibility of the 1,4-dicarbonyl precursors.
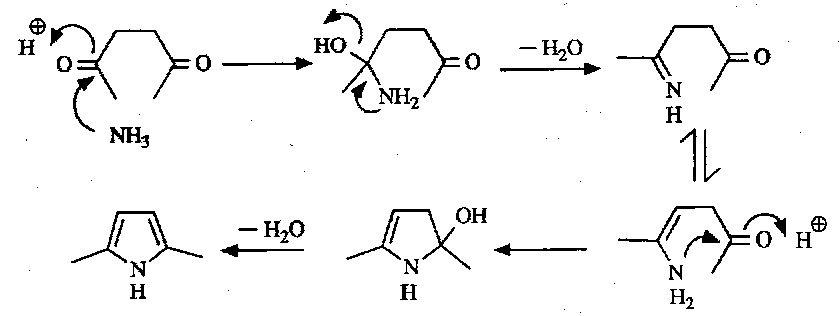
This synthesis can similarly be applied to thiophenes, e.g.

When H2S is used as the heteroatom source the mechanism is similar to the pyrrole above.
With Phosphorus(V) Sulphide, the situation is slightly different:

With furans, simple dehydration suffices to form the ring (i.e. no need to add one H2O molecule to then condense out two!).
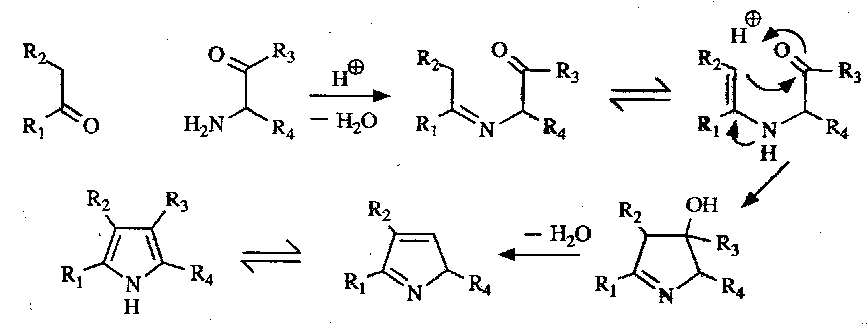
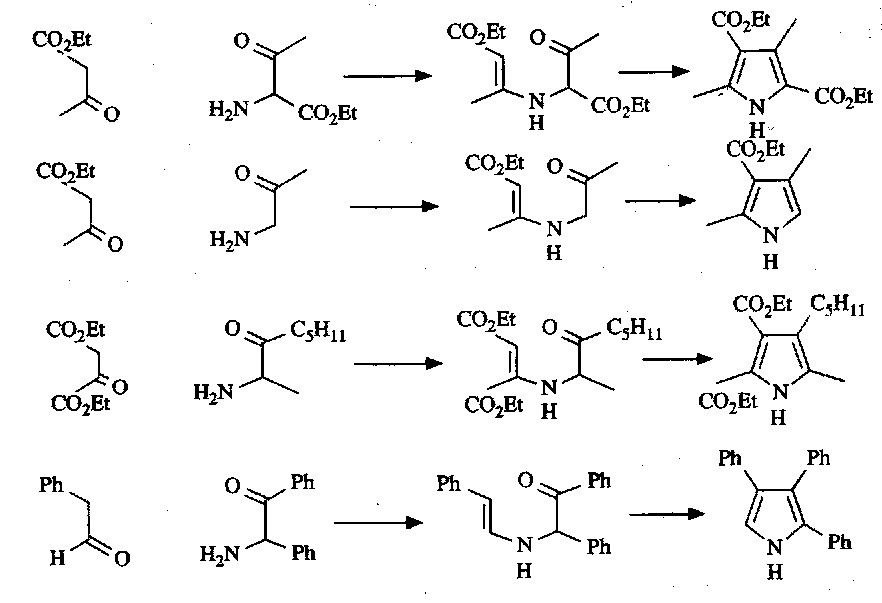
The most common method for pyrrole synthesis, however, is the Knorr Synthesis. This is the condensation of a ketone with an α-aminoketone via an enamine:
α-aminoketones can be prepared by nitrosation of an active methylene group followed by reduction of the oxime to the amine:
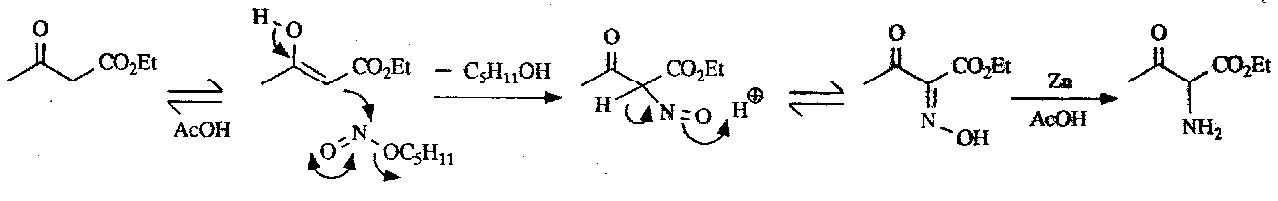
This is facilitated by R2 being electron-withdrawing (enhances the electrophilic nature of the ketone carbonyl and increases rate, preventing self-condensation).
A selection of the key intermediates / reagents for the Knorr Pyrrole Synthesis:
Electrophilic Substitutions
Generalised mechanism:
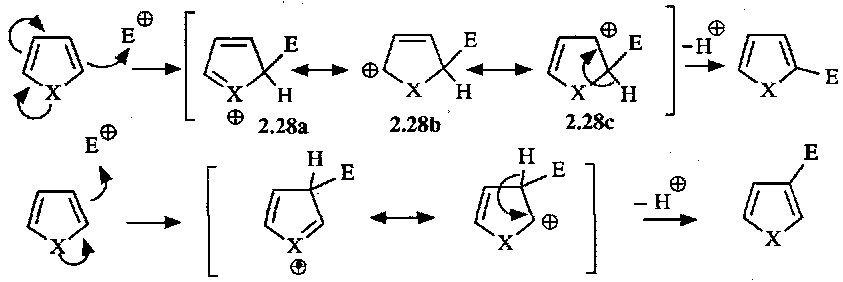
Ease of this reaction pyrrole > furan > thiophene > benzene. Reflects order of aromaticity (i.e. thiophene is more aromatic, so more stable to reaction).
Classic example of this reaction is the Vilsmeier Formylation of reactive aromatic compounds:
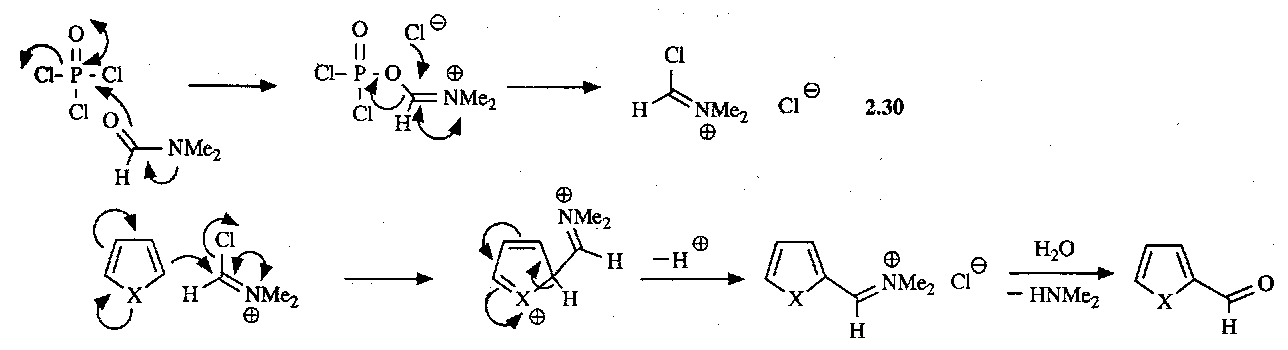
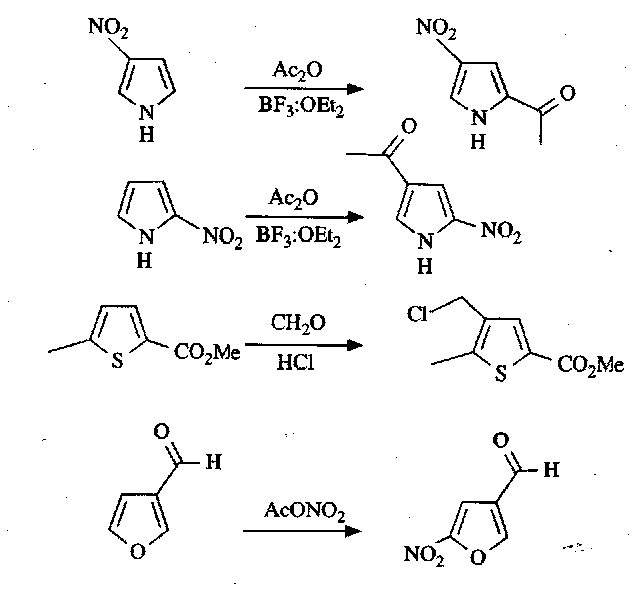
Many similar reactions can be performed, e.g. the Mannich Reaction. Friedel-Crafts can only be used on thiophenes, as pyrroles and furans are not stable to the Lewis Acid conditions required. However, the presence of Electron Withdrawing Groups on the ring stabilise them, so that it can be performed, e.g.
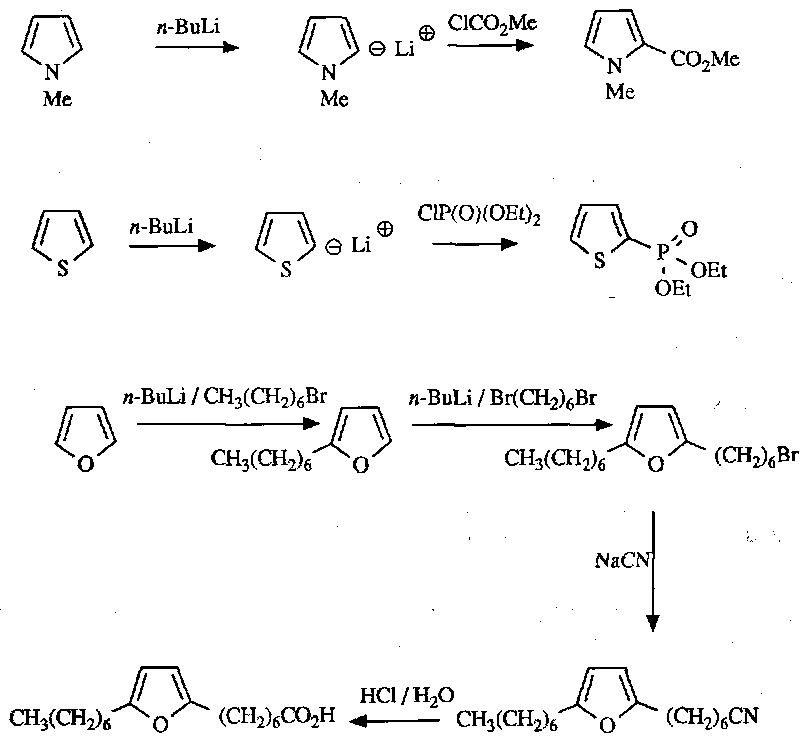
Anion Chemistry
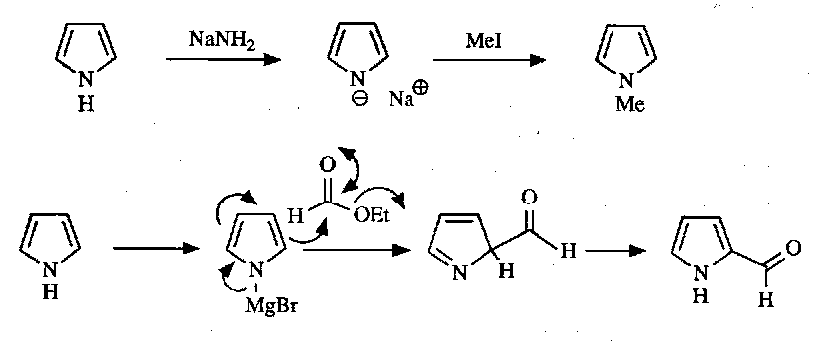
Useful reactions as a result:
Oxazoles, Imidazoles and Thiazoles
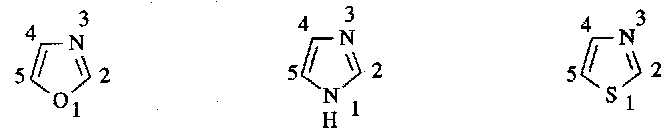
Synthesis
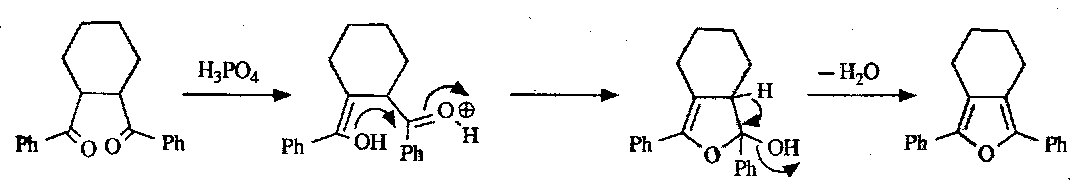
For oxazoles, the Robinson-Gabriel Synthesis is used:
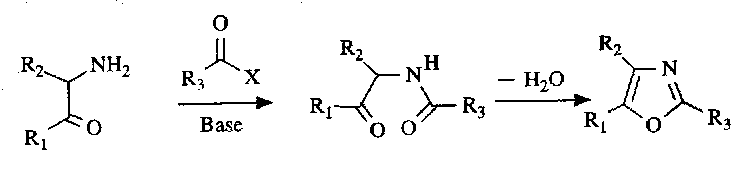
Dehydration can be carried out by a wide range of acids, such as phosphoric acid, phosgene or thionyl chloride. The mechanism is as follows:

There are several ways of preparing imidazoles. Condensation of a 1,2-dicarbonyl compound with ammonium acetate and an aldehyde is common:
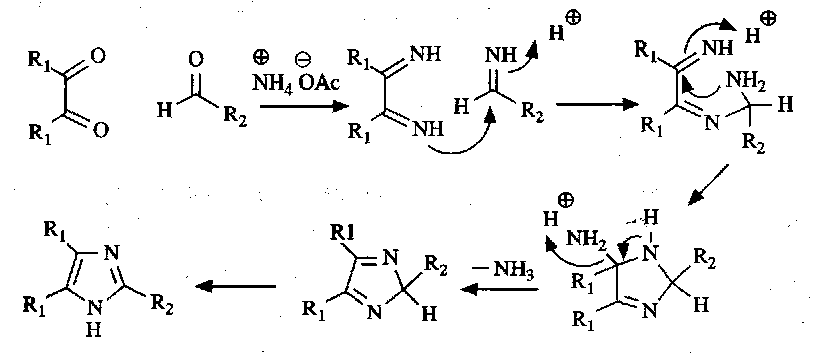
For thiazoles, the Hantzsch Synthesis is used:
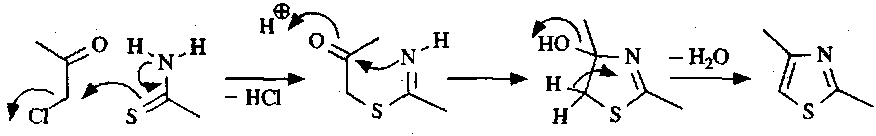
Electrophilic Substitution
1,3-azoles are not very reactive to electrophilic attack due to the deactivating effect of the pyridine-like Nitrogen. Electron-donating groups can facilitate this reaction though:
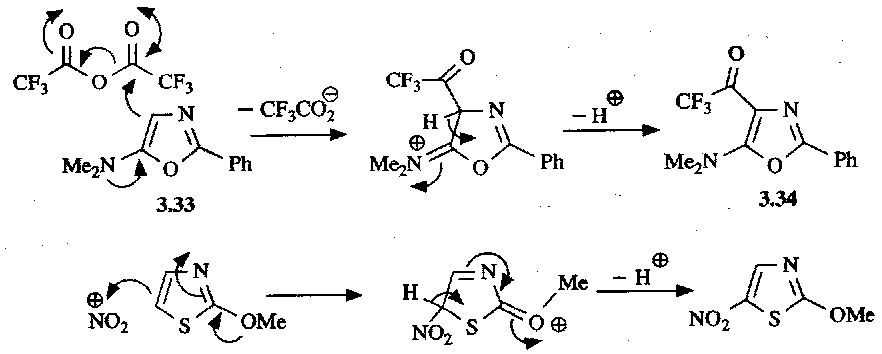
Anion Chemistry
The C2 position is particularly electron-deficient, so deprotonation here allows a wide variety of useful reactions to be carried out:
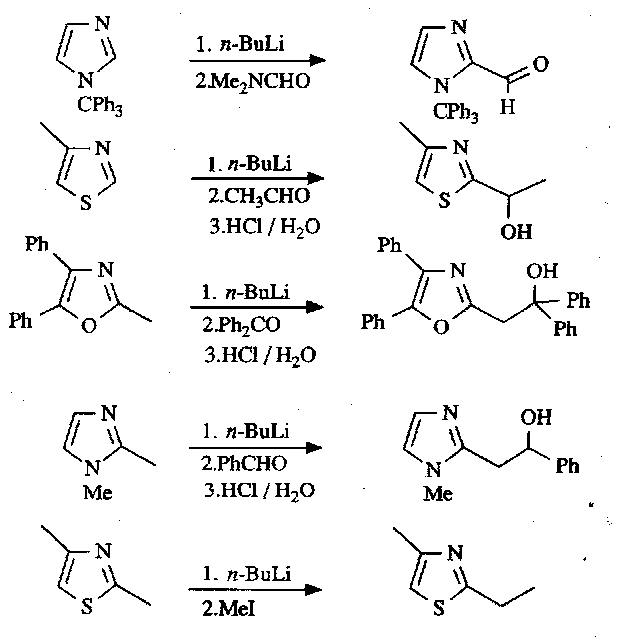
Nucleophilic Substitution
As a consequence of lower electrophilic reactivity, these are more reactive to nucleophiles. They require no activation with EWG like furans etc:
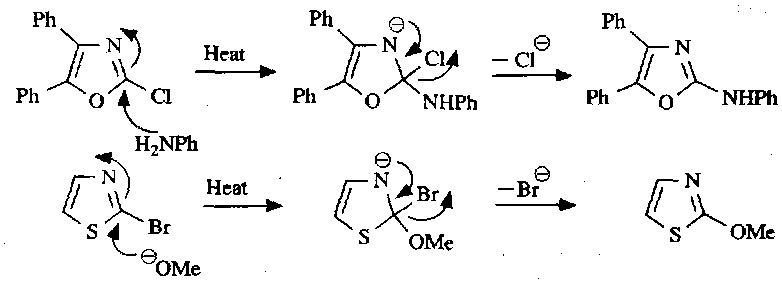
Isoxazoles, Pyrazoles and Isothiazoles
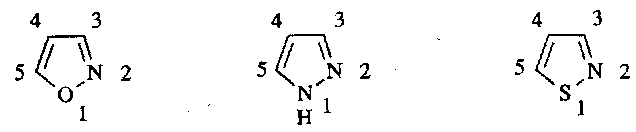
Synthesis
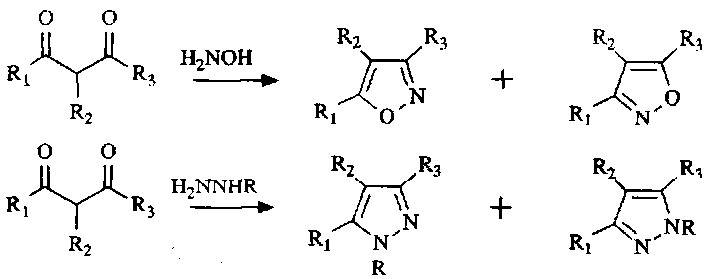
Isoxazoles and pyrazoles are synthesised in much the same way. The mechanism is:
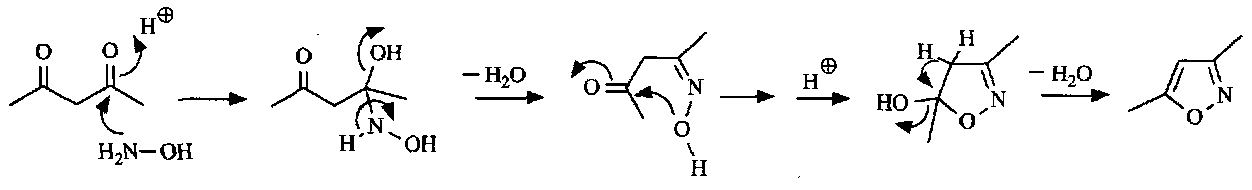
Hence, generalising,
Another important method of synthesis for isoxazoles involves [3+2] cycloaddition of nitrile oxides:
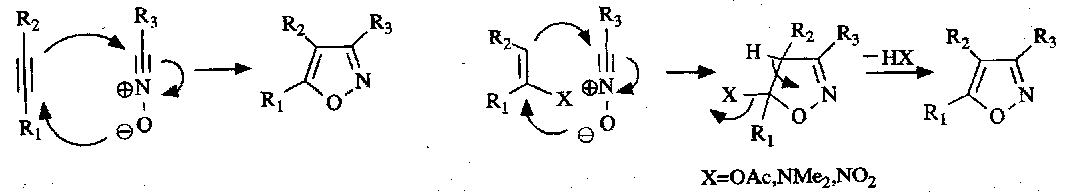
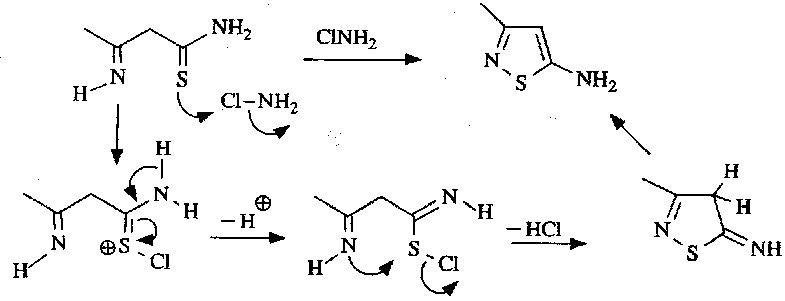
Isothiazoles are usually prepared by routes involving formation of the N-S bond in the cyclisation step. This is often set up by oxidation of the Sulphur atom, as in the following:
Electrophilic Substitution
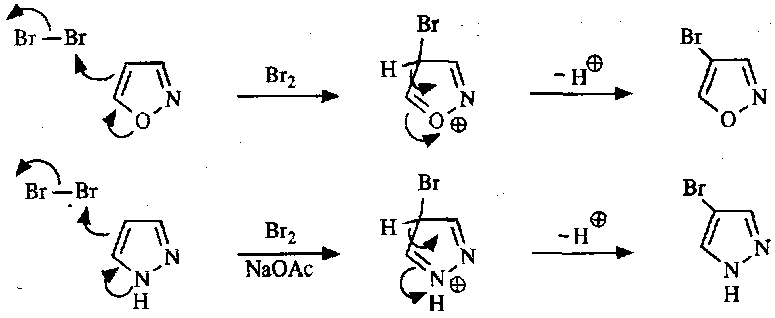
Less reactive than furan etc due to pyridine-like N present. It still occurs though, and principally at the C4 position, similar to the meta selectivity of pyridines.
Anion Chemistry
Deprotonate at C5 and then quenched with electrophiles, as previously:
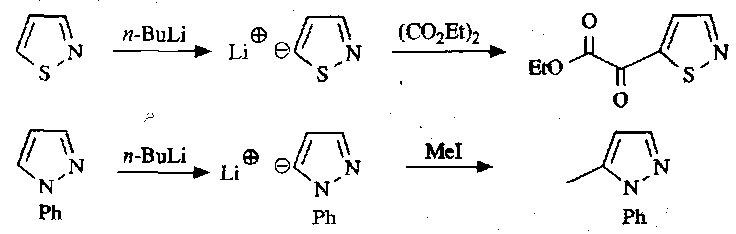
Note that this does not happen with isoxazoles, because the intermediate anion is unstable:
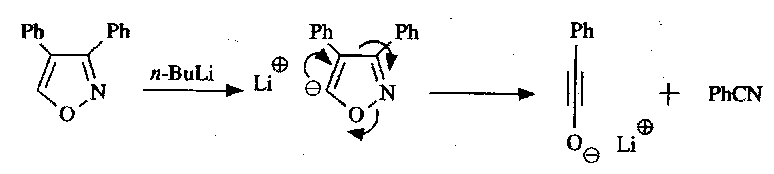
These can be deprotonated on attached alkyl groups however.
Pyridines
Synthesis
Retrosynthetically:
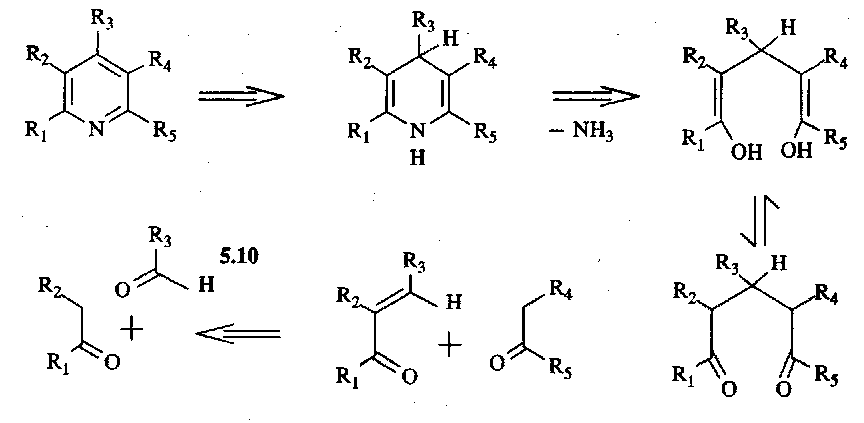
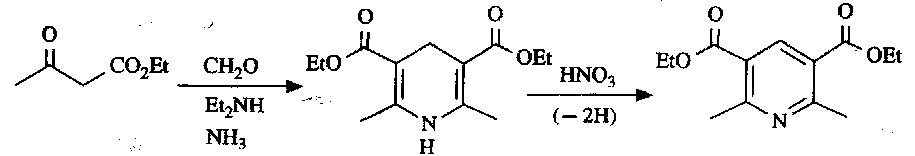
This gives rise to the classical Hantzch Pyridine Synthesis:
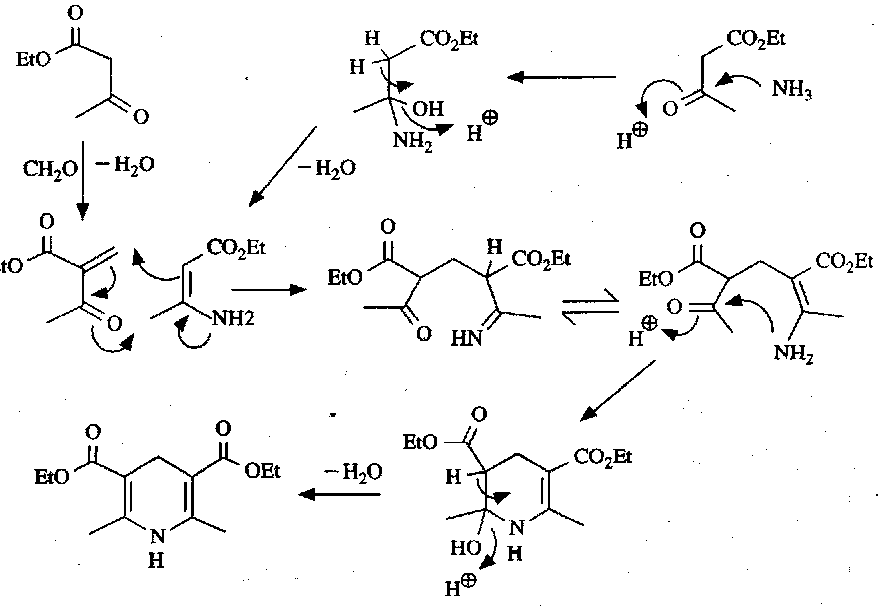
Mechanistically:
Electrophilic Substitution
Pyridine is virtually inert to aromatic electrophilic substitution. Its basic properties and the stabilisation conferred by the N atom mean that even nitration at the 3 position is arduous and low yielding.
However, pyridine can be activated by conversion to pyridine N-Oxide, and the O can subsequently be removed after the reaction is complete.

For example:
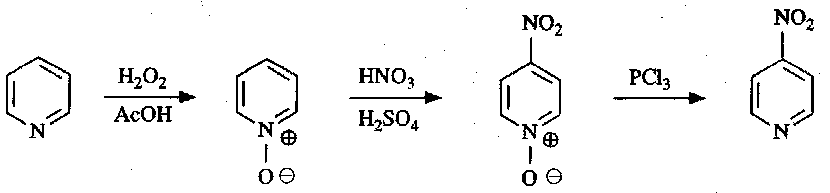
They can also be converted into synthetically useful 2-chloropyridines:

Another approach to electrophilic substitution involves the chemistry of 2-pyridone and 4-pyridone, which are tautomers of the hydroxypyridine.
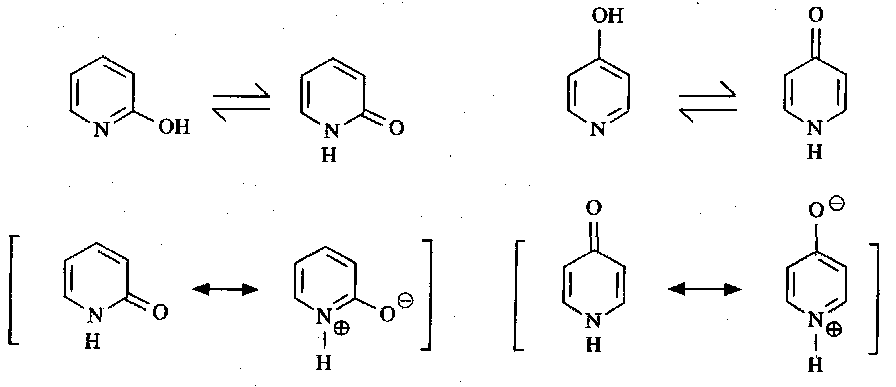
These react with electrophiles at the ortho and para positions to the activating O atom.
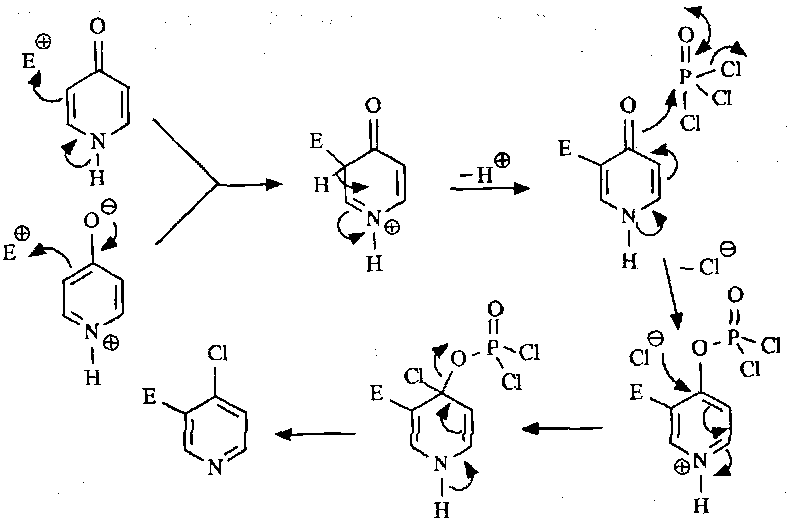
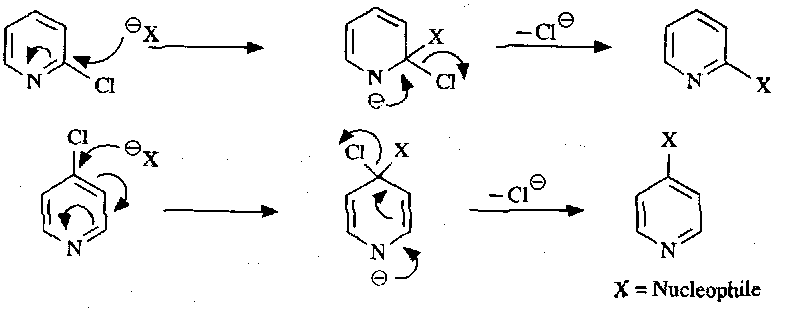
Nucleophilic Substitution
Pyridines can be attacked at the C2/C6 and the C4 positions:
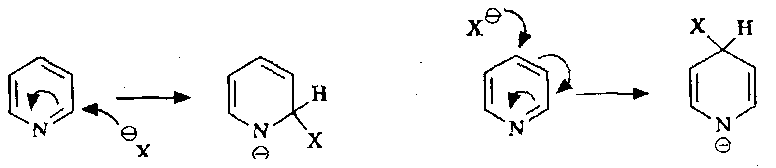
These are much more facile when better leaving groups are present:
Some example reactions:
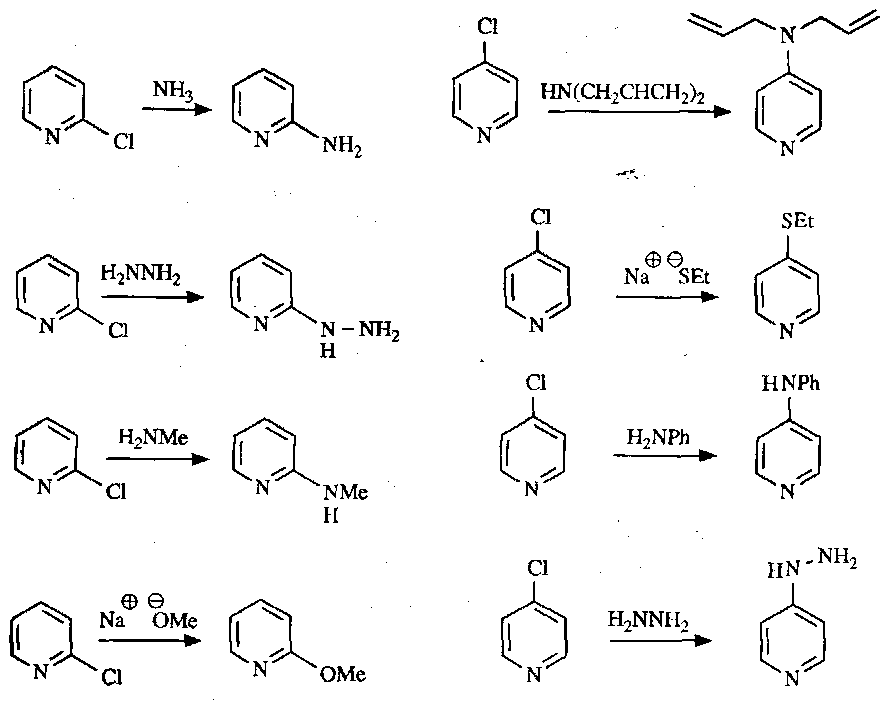
Anion Chemistry
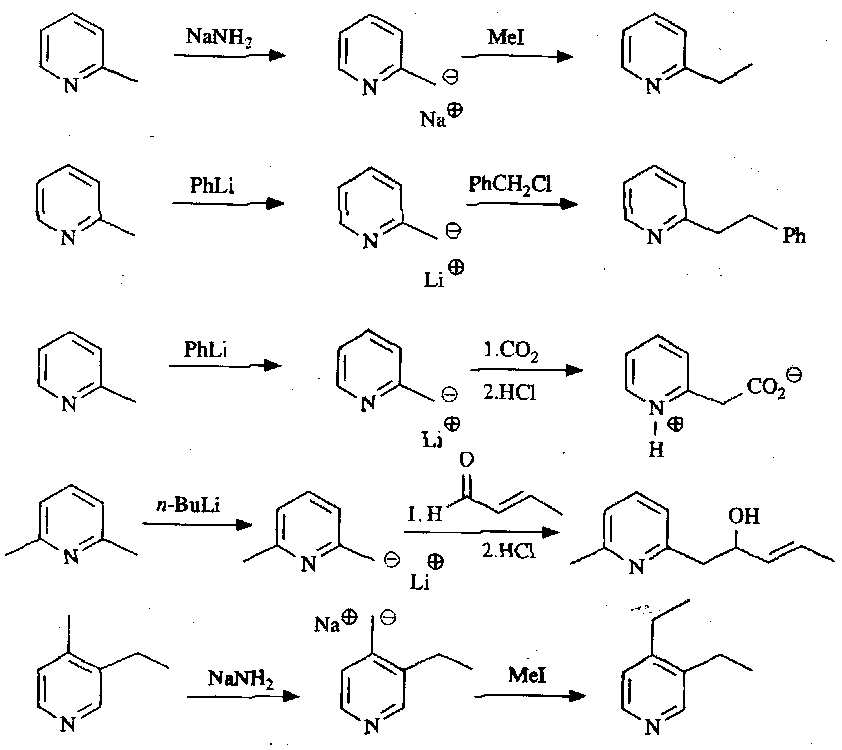
Deprotonation at C2/C6 and C4, for the same reasons as the nucleophilic attack locations. These then react with a range of nucleophiles:
Quinolines and Isoquinolines
Synthesis
The classic synthesis of quinolines is the Skraup Synthesis, which proceeds as follows:

The key intermediates in the synthesis of isoquinolines are β-arylethylamines. This is the Bischler-Napieralski Synthesis:
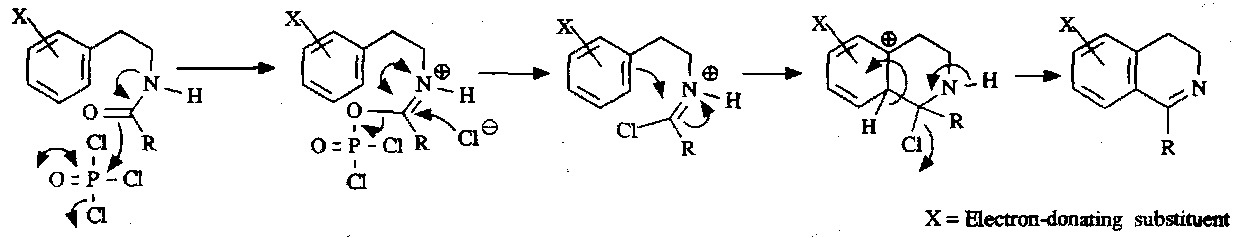
To achieve aromaticity from the product of this reaction, a Pd0 catalyst is used to dehydrogenate.
Electrophilic Substitution
These occur more easily than in pyridine, but the reaction occurs on the benzene ring as opposed to the pyridine.
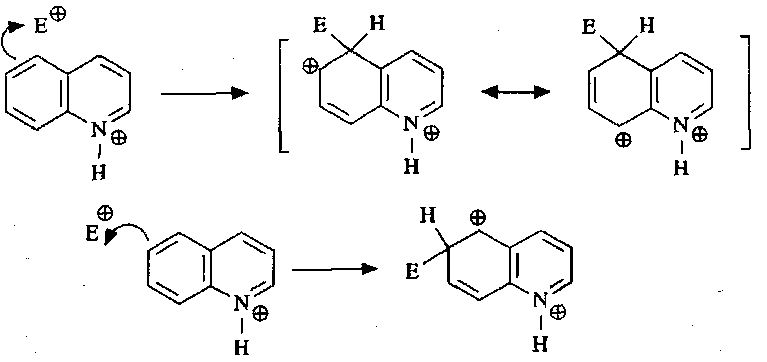
Nucleophilic Substitution and Anion Chemistry of quinolines are analogous to pyridines.
Indoles
Synthesis
Fischer Indole Synthesis:
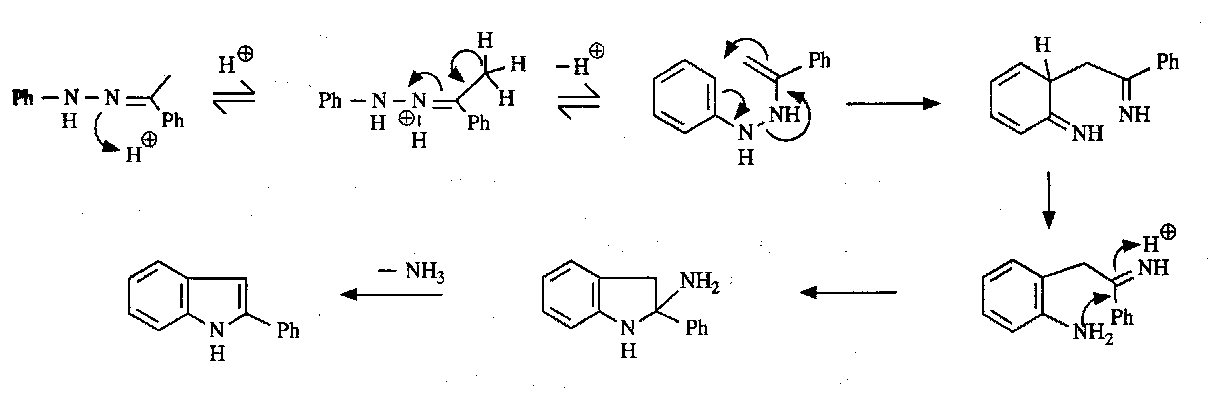
Note the pericyclic step in the mechanism.
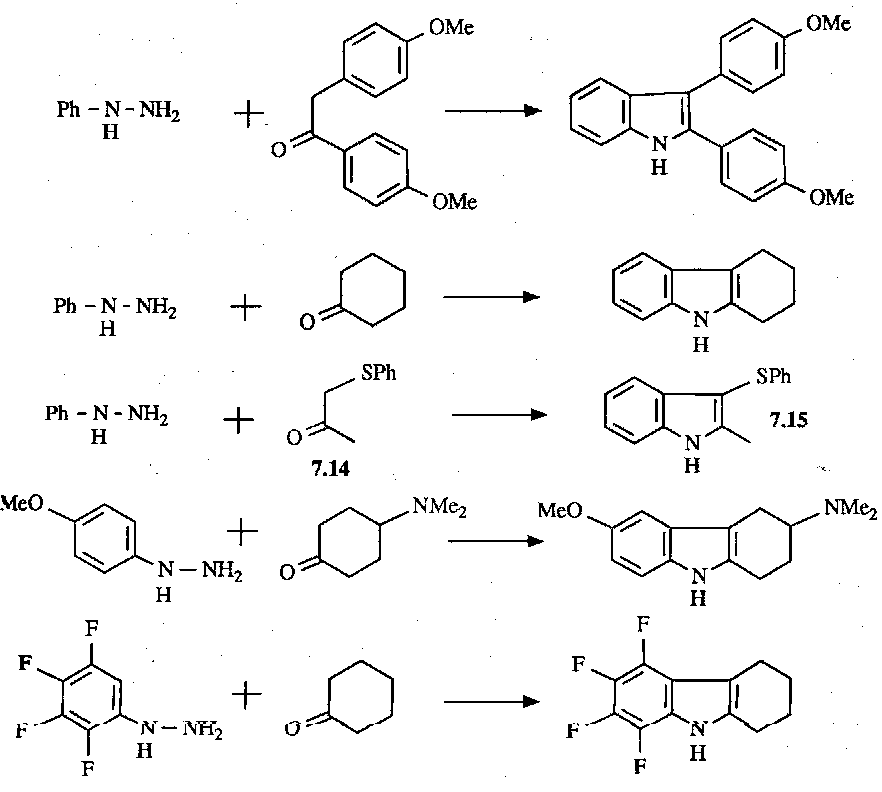
Some examples:
Electrophilic Substitution
This is facile in the electron-rich heterocycle, occurring preferentially at the C3 position.

Also, like pyrrole, Mannich-type reactions are easy:
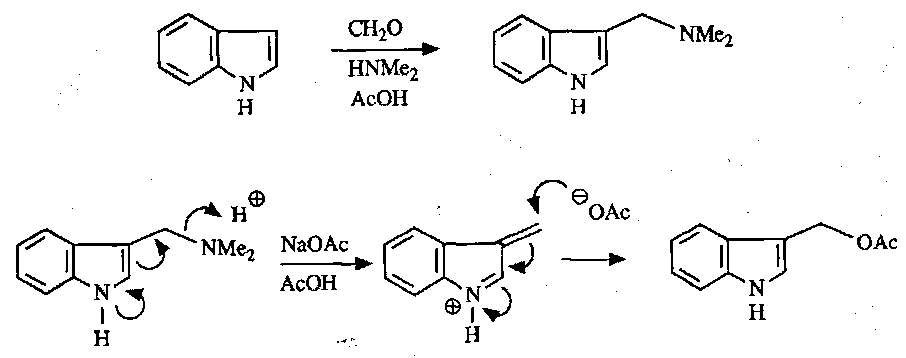
Vilsmeier Reaction is also suitable as above.
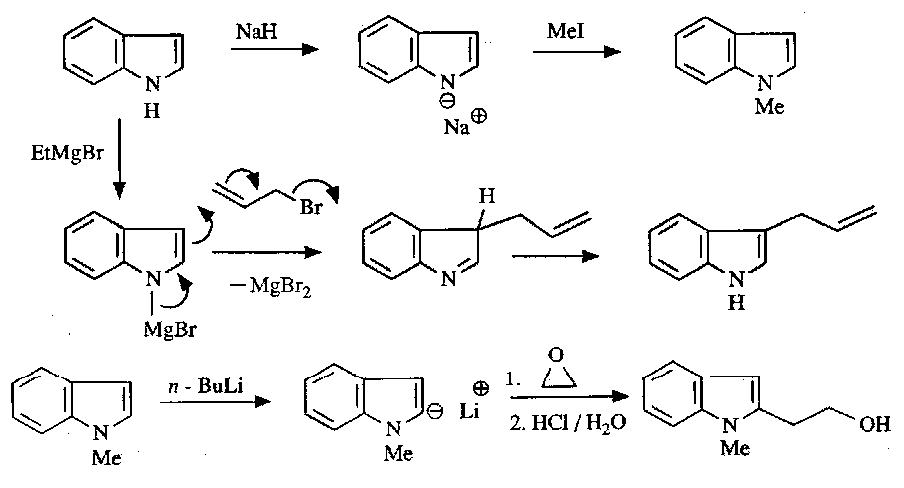
Anion Chemistry
Deprotonates at the N unless this is “protected” by reaction with an alkyl, in which case C2 is deprotonated.
I am happy for them to be reproduced, but please include credit to this website if you're putting them somewhere public please!